
Abstract
Automated microscopic analysis of stained histopathological images is degraded by the amount of color and intensity variations in data. This paper presents a novel unsupervised probabilistic approach by integrating a convolutional neural network (CNN) and the Gaussian mixture model (GMM) in a unified framework, which jointly optimizes the modeling and normalizing the color and intensity of hematoxylin- and eosin-stained (H&E) histological images. In contrast to conventional GMM-based methods that are applied only on the color distribution of data for stain color normalization, our proposal learns how to cluster the tissue structures according to their shape and appearance and simultaneously fits a multivariate GMM to the data. This approach is more robust than standard GMM in the presence of strong staining variations because fitting the GMM is conditioned on the appearance of tissue structures in the density channel of an image. Performing a gradient descent optimization in an end-to-end learning, the network learns to maximize the log-likelihood of data given estimated parameters of multivariate Gaussian distributions. Our method does not need ground truth, shape and color assumptions of image contents or manual tuning of parameters and thresholds which makes it applicable to a wide range of histopathological images. Experiments show that our proposed method outperforms the state-of-the-art algorithms in terms of achieving a higher color constancy.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
- Computational pathology
- Stain-color normalization
- Gaussian mixture model (GMM)
- Convolutional neural network (CNN)
1 Introduction
Computational histopathology involves computer-aided diagnosis (CAD) for microscopic analysis of stained histopathological slides to study presence, localization or grading of disease. Manual staining by adding contrasting dyes prior to microscopic imaging is a common clinical practice. Such a non-quantified manual procedure causes significant variations in tissue intensity and color, which also originate from other factors such as imaging device characteristics. Such undesirable effects become more problematic when different laboratories share digital images [1]. A well-known approach for compensating the color variations is applying color normalization techniques. Recent studies show that color normalization as preprocessing stage leads to a higher performance in a CAD system [2]. To do so, several methods are investigated [1, 3,4,5,6,7,8,9]. The proposed approaches can be divided into two categories:stain-color deconvolution and template color matching, which are first briefly explained below, and then we describe our contributions.
Stain-color deconvolution [10] methods are considering prior knowledge of the reference stain vector for every dye and split an input RGB image into three stain channels, each representing the actual stain color. Ruifrok et al. [10] introduce this prior knowledge by manually selecting pixels that represent a specific stain class and then compute the color deconvolution vector. Because of the semi-automatic nature, several studies are performed later for automated extraction of stains by e.g. using the singular value decomposition (SVD) technique [6], probabilistic Gaussian mixture model (GMM) [8], using a prior for stain matrix estimation [3] and stain color descriptions along with training a supervised relevance vector machine [4]. Although these solutions lead to a better stain estimation, they solely limited to image color information, while the spatial dependency among tissue structures has been ignored [1]. This causes shortcomings for stain deconvolution approaches when severe staining variations occur in the data.
Template color matching methods proposed by Reinhard et al. [5] rely on aligning the statistical color distribution (e.g. mean and standard deviation) of a source image with a template image. The authors used a set of linear transformations for assigning a unimodal distribution to each channel of the CIELAB color model. Afterwards, each channel was treated independently for alignment. Since there is a dependency between the color channels due to dye contribution, this approach has drawbacks which have been addressed in [1, 4]. For solving this problem, separate transformations are performed on stain classes [8], or on tissue classes [1, 9]. For avoiding artifacts at the border of different classes under different transformations, a weighted contribution of these transformations in the final color image is considered. Two proposed solutions consist of estimating weights of the GMM [8] and training a naive Bayesian classifier [1]. This solution introduce multiple parameters and thresholds which cannot be optimally applied to a new dataset or even fail if the tissue type changes.
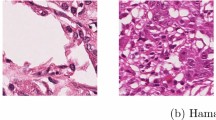
Regarding to [8, 9], we propose a stain-color normalization method based on GMM but with a different approach. Our contribution is threefold. First, we introduce a new unsupervised stain-color normalization method based on deep convolutional Gaussian mixture models (DCGMM). Our proposal benefits from a convolutional neural network (CNN) for performing soft-assignment clustering of tissue structures in an unsupervised manner. Second, in contrast to the previous GMM-based color normalization methods that work on color point-clouds by considering each pixel independently in input space, our method fits a GMM to an input color image with processing and involving the visual contents, such as the appearance and shape of regions in the image intensity (density) channel by means of the CNN. Such an approach outperforms the previous methods specifically when strong staining variations appear in the data (see Fig. 1). This outcome occurs because our method is independent from the chromatic information for tissue class assignment and consequent distribution modeling. Finally, instead of using a common expectation-maximization (EM) algorithm for optimizing the GMM, we introduce an end-to-end learning procedure for optimizing the parameters of the CNN and the GMM together. To our knowledge, it is the first time that this approach is used for training a DCGMM.

Example of standard GMM failure in tissue clustering using HSD color space; (a) RGB H&E image; (b) 3-class standard GMM; (c) 3-class DCGMM.
2 Methods
We first provide a brief overview of the standard GMM method and introduce the notation used in the paper. Afterwards, we introduce our DCGMM method.
Gaussian Mixture Model of data (\(\mathbf {x}\)), can be presented as a linear superposition of K Gaussian mixtures in terms of discrete latent variables (\(\mathbf {z}\)), in the form of
The K-dimensional binary random variable \(\mathbf {z}\) has one-hot encoding (\(z_k\in \{0,1\}\); \(\varSigma ^K_{k=1}z_k = 1\)), which represents the tissue class in our study. In Eq. (1), the mixing coefficients \(\pi _k\) must satisfy \(0\le \pi _k\le 1\) together with \(\varSigma ^K_{k=1}\pi _k = 1\), in order to fulfill a valid probability definition [11]. Here, \(\mathcal {N}\) stands for a multivariate normal distribution with mean \(\varvec{\mu }_k\) and covariance matrix \(\mathbf {\Sigma }_k\). If we consider \(\pi _k\) as prior probability of class \(z_k\), its posterior probability called responsibility can be written as follows [11]:
According to Eq. (1), the (natural) log-likelihood function for an image (\(\mathbf {X}=\big \{\mathbf {x}_1,\mathbf {x}_2,...,\mathbf {x}_N\big \}\)) with total number of pixels (observations) equal to N is
Given the GMM, the goal is to maximize the likelihood function (Eq. (3)) with respect to the parameters (\(\varvec{\mu }_k\), \(\mathbf {\Sigma }_k\), \(\pi _k\)). The common approach for this is using the EM algorithm by iteratively evaluating the responsibilities (Eq. (2)) and re-estimating the parameters.

Block diagram of DCGMM in training phase.
Deep Convolutional GMM (DCGMM): The recent development of deep generative models has invoked some extensions to the standard GMM [12, 13]. Two proposed approaches are (1) constructing a stack of multiple GMM layers on top of each other in a hierarchical architecture [13] that is optimized by EM-based algorithm and (2) using auto-encoder neural networks while applying the GMM on their low-dimensional representations [12] that has been studied for unsupervised anomaly detection. This paper presents a different extension to the standard GMM by introducing the DCGMM that is a fully-convolutional CNN of which the parameters are optimized to fit a GMM to the input image, in an end-to-end learning, using gradient descent and back-propagation algorithms.
We aim to fit a GMM to the pixel-color distribution conditioned on tissue classes. For processing the image and detecting the tissue classes, the high capability of the CNN has been exploited. To do so, estimating the responsibility coefficients (Eq. (2)) is performed by a CNN. For a better understanding, one can consider that the E-step in an EM-based optimization is replaced by a CNN. However, all parameters of the GMM and CNN are jointly optimized by the gradient descent algorithm. In the DCGMM, the negative log-likelihood (maximizing Eq. (3)) is used as the loss function.
Our color normalization algorithm can be split into two phases, training the DCGMM and the color transformation (inference). In the training phase, we fit a GMM to the data. After the training stage is finished and in inference mode, the template image and source image are separately supplied to the model. Consequently, the parameters of the fitted Gaussian distributions and their mixture coefficients (\(\varvec{\pi }\)) are computed in those two images. Afterwards, the multivariate Gaussian distributions of the source image are transformed (aligned) to have similar parameters of distributions as the template image, while \(\varvec{\pi }\) is kept unchanged. In the rest of this section, we explain these two phases in detail.
Fitting Gaussian Distribution to Data: First the RGB color values are transferred to the hue-saturation-density (HSD) color system [14]. In a HSD model of histopathological images, the density channel (D) is linearly related to the amount of stain while the other two chromatic channels are independent. This property suits well to the analysis of transmitted light microscopy, compared to the alternative color spaces [1]. We only use the normalized to zero-mean (centered) density channel as the input to the network. Ignoring the chromatic information and only clustering the tissue structures according to their appearance (normalized density channel), alleviating the effect of strong staining variations in images. The proposed CNN has a fully-convolutional architecture, consisting of several convolutional layers, rectified linear units (ReLU) as nonlinearity functions and (un)pooling operators. The reduced image size after applying two stages of max-pooling returns back to it original size by applying un-pooling operations. After the last convolutional layer, there is a softmax layer. The network aims to estimate the responsibility values (see Eq. (2)) for each pixel in the input image. Using the softmax layer at the output of the network guarantees the constraint of \(\varSigma ^K_{k=1}\gamma _k = 1\). Since we are working on H&E histopathological images, each pixel in the image mostly belongs to one out of three clusters (\(K=3\)): hematoxylin, eosin and background (not stained). Because the biological composition of tissue related to each pixel in the image leads to a varying stain absorption ratio between pixels, the color of each pixel can be presented by a weighted sum of the different stains used. This property can be reflected in the responsibility coefficient (\(\varvec{\gamma }\)) of a GMM, which is estimated in the softmax layer of the network in our proposed model. The calculation of the required partial derivatives of negative log-likelihood (loss function) with respect to its parameters (\(\varvec{\pi },\varvec{\mu }\) and \(\varvec{\varSigma }\)) for performing a gradient descent algorithm can be found in [15, p. 45].

Although the responsibility term is estimated by the CNN from only the density channel of the image, the mean and covariance matrices (\(\varvec{\mu }_k\) and \(\mathbf {\Sigma }_k\)) are estimated from all three channels of the HSD color image. The randomly initialized parameters of the CNN are updated by ADAM gradient-based optimization with a fixed learning rate of \(10^{-3}\). The scheme of our model is depicted in Fig. 2.
Transformation of Multivariate Gaussian Distributions: By training the model in the color normalization task, two GMMs are fitted to the source and template images, individually. Afterwards, a set of transformations are applied to align the multivariate Gaussian distributions between the source and the template. These transformations consist of three operations: mean centering, whitening and coloring transformation. Let us assume that (\(\varvec{\mu }^s,\mathbf {\Sigma }^s\)) and (\(\varvec{\mu }^t,\mathbf {\Sigma }^t\)) are the estimated parameters of the two distributions in source and template image, respectively. By shifting the mean of source image to the origin (centering) and then whitening which involves the SVD algorithm, the source image will have a zero mean and an identity covariance matrix. Consequently, by applying a coloring transformation which is the inverse of whitening but after replacing the eigenvalues (\(\varvec{\varLambda }\)) and eigenvectors (\(\varvec{\varPhi }\)) of source distribution with the template distribution, the whitened Gaussian distribution of source image scales and rotates to have the same covariance matrix as the template image (\(\mathbf {\Sigma }^t\)). Finally, the distribution is shifted to obtain the same mean as the template distribution. For clarifying this procedure, pseudocode in Algorithm 1 shows these steps in detail.
3 Experimental Results
Histopathology Image Dataset: We focus on inter-laboratory variations of the H&E staining, as it is a major concern in large-scale application of CAD in pathology. For better comparison with recent studies, we use the same dataset as has been introduced in [1]. The dataset contains 625 images (each \(1388\times 1040\) pixels) from 125 digitized H&E stained WSIs of lymph nodes from 3 patients and was collected from five Dutch pathology laboratories, each using their own routine staining protocols (more details can be found in [1]). Our model is trained on randomly cropped patches (\(576\times 576\) pixels) and evaluated on full-sized images by using leave-one-out cross-validation based on the above laboratories.
Results: We trained the DCGMM on the dataset. The model easily converges in a few minutes. The inference computation time for each image in its original size (\(1388\times 1040\) pixels) is about 0.6 s, implemented in the TensorFlow library and running on a TITAN Xp GPU. The performance of our method is compared to that of five competing state-of-the-art algorithms: linear appearance normalization by Macenko et al. [6], statistical color properties alignment by Reinhard et al. [5], nonlinear mapping for stain normalization by Khan et al. [4], sparse non-negative matrix factorization by Vahadaneet al. [7] and WSI color standardizer by Bejnordi et al. [1]. The normalized median intensity (NMI) measure [1, 9] is used to evaluate color constancy of normalized images. Quantitative analysis is based on independently evaluating the color constancy in the regions that show mostly absorbed hematoxylin or eosin. Since nuclei mostly absorb hematoxylin, they first are detected automatically by using a fast radial symmetry transform and a marker-controlled watershed algorithm [1]. Since our generated hematoxylin masks slightly differ from what were used in [1], the obtained NMI scores in our benchmark are not exactly the same as [1]. However, the results are in agreement with [1]. For evaluation of the eosin analysis, several regions are manually annotated for 25 images. The evaluation results of different methods for assessing hematoxylin and eosin regions are shown in Table 1. The results clearly indicate that our proposed method results in the lowest variation in color after normalization of the images and it outperforms competing state-of-the-art methods. Figure 3 illustrates an example of the template image, a source image and the outcomes of color normalization by the different methods.

4 Conclusions
We have introduced a framework for modeling and normalizing the colors in H&E histopathological images. Our model jointly optimizes the parameters of a CNN and the parameters of multivariate GMM in an end-to-end learning framework. By minimizing the negative log-likelihood loss function, the CNN learns how to cluster the image structures for optimally fitting the GMM on the data. Our proposal takes only one assumption on the number of clusters that is evidently chosen to three (\(K=3\)) for H&E images. It does not need manual tuning of parameters and thresholds which makes it applicable to a wide range of histopathological images collected from different organs. Since our method processes and fits the GMM conditioned on the tissue classes, in comparison with previous methods, it is more robust in presence of strong stain variations.
References
Bejnordi, B., et al.: Stain specific standardization of whole-slide histopathological images. IEEE Trans. Med. Imaging 35(2), 404–415 (2016)
Ciompi, F., Geessink, O., Bejnordi, B.E., et al.: The importance of stain normalization in colorectal tissue classification with convolutional networks. In: 2017 IEEE 14th International Symposium on Biomedical Imaging (ISBI 2017), pp. 160–163. IEEE (2017)
Niethammer, M., Borland, D., Marron, J.S., Woosley, J., Thomas, N.E.: Appearance normalization of histology slides. In: Wang, F., Yan, P., Suzuki, K., Shen, D. (eds.) MLMI 2010. LNCS, vol. 6357, pp. 58–66. Springer, Heidelberg (2010). https://doi.org/10.1007/978-3-642-15948-0_8
Khan, A., Rajpoot, N., Treanor, D., Magee, D.: A nonlinear mapping approach to stain normalization in digital histopathology images using image-specific color deconvolution. IEEE Trans. Biomed. Eng. 61(6), 1729–1738 (2014)
Reinhard, E., Adhikhmin, M., Gooch, B., Shirley, P.: Color transfer between images. IEEE Comput. Graph. Appl. 21(5), 34–41 (2001)
Macenko, M., et al.: A method for normalizing histology slides for quantitative analysis. In: Proceedings of IEEE International Symposium on Biomedical Imaging Nano Macro, pp. 1107–1110 (2009)
Vahadane, A., et al.: Structure-preserving color normalization and sparse stain separation for histological images. IEEE Trans. Med. Imaging 35(8), 1962–1971 (2016)
Magee, D., et al.: Color normalization in digital histopathology images. In: Proceedings of Optical Tissue Image Analysis Microscopy, Histopathology Endoscopy, pp. 100–111 (2009)
Ajay Basavanhally, A.M.: Em-based segmentation-driven color standardization of digitized histopathology. Proc. SPIE 12, 8676–8676-12 (2013)
Ruifrok, A.C., Johnston, D.A.: Quantification of histochemical staining by color deconvolution. Analyt. Quant. Cytol. Histol. Int. Acad. Cytol. Am. Soc. Cytol. 23(4), 291–299 (2001)
Christopher, M.B.: Pattern Recognition and Machine learning. Springer, New York (2016)
Zong, B., et al.: Deep autoencoding gaussian mixture model for unsupervised anomaly detection. In: International Conference on Learning Representations (2018)
van den Oord, A., Schrauwen, B.: Factoring variations in natural images with deep gaussian mixture models. In: Advances in Neural Information Processing Systems, vol. 27, pp. 3518–3526. Curran Associates, Inc. (2014)
van der Laak, J.A., Pahlplatz, M.M., Hanselaar, A.G., de Wilde, P.C.: Hue-saturation-density (hsd) model for stain recognition in digital images from transmitted light microscopy. Cytometry 39(4), 275–284 (2000)
Petersen, K.B., Pedersen, M.S.: The Matrix Cookbook. Technical University of Denmark, version 20121115, November 2012
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this paper

Cite this paper
Ghazvinian Zanjani, F., Zinger, S., de With, P.H.N. (2018). Deep Convolutional Gaussian Mixture Model for Stain-Color Normalization of Histopathological Images. In: Frangi, A., Schnabel, J., Davatzikos, C., Alberola-López, C., Fichtinger, G. (eds) Medical Image Computing and Computer Assisted Intervention – MICCAI 2018. MICCAI 2018. Lecture Notes in Computer Science(), vol 11071. Springer, Cham. https://doi.org/10.1007/978-3-030-00934-2_31
Download citation
DOI: https://doi.org/10.1007/978-3-030-00934-2_31
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-00933-5
Online ISBN: 978-3-030-00934-2
eBook Packages: Computer ScienceComputer Science (R0)