
Abstract
We propose a cancer grading approach for transrectal ultrasound-guided prostate biopsy based on analysis of temporal ultrasound signals. Histopathological grading of prostate cancer reports the statistics of cancer distribution in a biopsy core. We propose a coarse-to-fine classification approach, similar to histopathology reporting, that uses statistical analysis and deep learning to determine the distribution of aggressive cancer in ultrasound image regions surrounding a biopsy target. Our approach consists of two steps; in the first step, we learn high-level latent features that maximally differentiate benign from cancerous tissue. In the second step, we model the statistical distribution of prostate cancer grades in the space of latent features. In a study with 197 biopsy cores from 132 subjects, our approach can effectively separate clinically significant disease from low-grade tumors and benign tissue. Further, we achieve the area under the curve of 0.8 for separating aggressive cancer from benign tissue in large tumors.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Prostate Cancer (PCa) is a significant public health issue. According to the National Cancer Institute (NCI)Footnote 1, approximately 14 % of men will be diagnosed with PCa at some point during their lifetime. Definitive diagnosis involves core needle biopsy guided by Transrectal Ultrasound (TRUS), followed by histopathological analysis of the obtained samples. TRUS is blind to intraprostatic pathology, and can miss clinically significant disease [5].
In recent years, multi-parametric Magnetic Resonance Imaging (mp-MRI) and its fusion with TRUS has emerged as a promising technology to target potential cancer lesions identified in mp-MRI [13, 14]. While mp-MRI has negative predictive values as high as 94 % [9, 12], it has a high false positive rate, and can miss smaller tumors. Furthermore, mp-MRI can not reliably detect the degree of aggressiveness of cancer, known as the grade. The vagaries of PCa diagnosis and prognosis have led to high rates of over-treatment: for every man saved from PCa-related death, 1400 are screened and 48 undergo radical treatment [8]. Accurate detection of aggressive cancer is critical to its appropriate management. Patients with indolent cancer can then opt for active surveillance [5, 13].
There have been a large number of efforts to adopt ultrasound (US)-based tissue typing for PCa detection as the US is affordable, accessible and real time. PCa detection using analysis of B-mode images [10] and single frame radio-frequency (RF) US data [6] has not had significant clinical uptake, while the application of these methods for PCa grading is not widely reported. Elastography [3] and Doppler imaging [11], available on many conventional US systems, have been promising for PCa detection while conflicting results have been reported on their application to PCa grading [8]. The main shortcoming of these approaches is the need to determine a consistent threshold for tissue properties that can reliably identify cancer, and generalize well to prospective patients [3].
More recently, analysis of temporal US data has emerged as a promising modality for PCa tissue typing. In this technology, a series of US frames is captured from a stationary tissue location without intentional movement of the tissue or the transducer. This approach has been successful in classification of cancerous and benign prostate tissue [1, 7]. It has also been employed to differentiate between various cancer grades, in preliminary whole-mount studies [8].
In this paper, in a clinical study of 197 TRUS-guided biopsy cores from 132 patients, we use temporal US to address the problem of PCa grading. We propose an approach that is based on deep learning and statistical analysis of image regions corresponding to biopsy targets. It has two components (Fig. 1): (1) feature learning, where a deep learning architecture derives a set of high-level latent features to separate benign from the cancerous tissue; and (2) distribution learning, where clustering is applied in the space of the latent features to determine the cancer grade. Our proposed approach is effective in differentiating aggressive PCa from clinically-less-significant disease and non-cancerous tissue.
2 Materials and Methods
2.1 Data
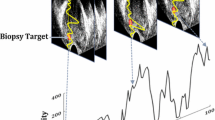
One hundred and thirty-two (132) subjects were enrolled in the study. All subjects provided informed consent to participate and the study was approved by the institutional research ethics board. The subjects underwent a diagnostic mp-MRI of the prostate. The mp-MRI sequences were examined by two independent radiologists to identify primary and secondary cancerous lesions (with cancer suspicious level assigned to as low, intermediate, or high), and to provide the “largest diameter of tumor”. Subjects with suspicious lesions underwent MRI-guided targeted TRUS biopsies using the UroNav (Invivo Corp., FL) MR/US fusion system. During biopsy, T2-weighted MR images were registered to the 3D US volume of the prostate using UroNav. The clinician then navigated in the prostate volume towards the MR-identified target; the TRUS transducer was held steady for about 5 s to acquire 100 frames of temporal US data from the target, and the biopsy core was taken. Two cores were obtained for the primary lesion; one in the axial, the other in the sagittal plane. Temporal US data was only recorded from the primary lesion in the axial imaging plane to minimize disruption of the clinical work flow. Histopathology labels of the cores were used as the ground-truth (Fig. 1).

An illustration of the proposed cancer grading approach.
For each target, the Gleason Score (GS) and the % distribution of PCa in the axial and sagittal samples were reported. The GS is used to describe PCa grade and ranges from 1 (resembling normal tissue) to 5 (aggressive cancerous tissue). It is reported as a sum of the grades of the two most common patterns in a tissue specimen. We only include cores in our study where the axial and sagittal pathology match. From 197 cores in our data, 57 were cancerous (12 GS of 3 + 3, 19 GS of 3 + 4, four GS of 4 + 3, 20 GS of 4 + 4, and two GS of 4 + 5) while 140 had non-cancerous histology including benign or fibromuscular tissue, chronic inflammation, atrophy and Prostatic Intraepithelial Neoplasia (PIN). We divide the data from 197 cores into training and testing sets. Training data consists of 32 biopsy cores from 27 patients with the following histopathology labels: 19 benign, 0 GS 3+3, 5 GS 3+4, 2 GS 4+3, 4 GS 4+4 and, 2 GS 4+5. The test data is made up of 165 cores from 114 patients, with the following distribution: 121 benign, 12 GS 3+3, 14 GS 3+4, 2 GS 4+3, and 16 GS 4+4.
2.2 Preprocessing
We compute the spectrum of temporal US data obtained from each biopsy core. For this purpose, we analyze an area of \(2\times 10\,\mathrm{mm}^2\) around the target location in the lateral and axial directions, respectively. This region is along the projected needle path in the US image and centered on the target. We divide the selected area to 20 equally-sized Regions of Interest (ROI) of size \(1\,\mathrm{mm}^2\). For each ROI, we take the Fourier transforms of all time series corresponding to the RF samples in each ROI, normalized to the frame rate. Then, we average the absolute values of the Fourier transforms of the RF time series in each ROI. Finally, each ROI is represented by 50 positive frequency components (see Fig. 1).
2.3 Cancer Grading
Grading can be considered as a multi-class classification problem, where the objective is to determine if an area in the tissue is benign or has various grades of PCa (Grades 3, 4 or 5). Training such a classifier with prostate biopsy data is non-trivial: the ground-truth histopathology reports a measure of the statistical distribution of cancer in a biopsy core. The exact location of the cancerous tissue in the core is not provided. Therefore, the exact label of each ROI in a core is not available, rather the statistics of ROIs with various labels in a core are known. We propose a coarse-to-fine classification approach that similar to histopathology reporting, calculates a statistical representation of the distribution of ROIs in various classes (benign and grades 3, or 4). The approach has two steps (Fig. 1): (1) feature learning to extract latent features that maximally separate benign from cancerous tissues; and (2) distribution learning to model the statistical distribution of cancer grades in the space of learnt features.
Feature Learning: We use a Deep Belief Network (DBN) structure [1] to map the set of 50 spectral components for each ROI to six high-level latent features. The network structure includes 100, 50 and 6 hidden units in three layers, where the last hidden layer represents the latent features. In the pre-training step, the learning rate is fixed at 0.001, mini-batch size is 5, and the epoch is 100. Momentum and weight cost are set to defaults of 0.9, and \(2 \times 10^{-4}\), respectively. For discriminative fine-tuning, a node is added to represent the labels of observations, and back-propagation with a learning rate of 0.01 for 70 epochs and mini-batch size of 10 is used. We perform dimensionality reduction in the space of the latent features. We use Zero-phase Component Analysis [2] to whiten the features and determine the top two eigen vectors, \(f_1\) and \(f_2\). We call this space the eigen feature space.
Distribution Learning: We use the training data to build a Gaussian Mixture Model (GMM) [15] to represent the distribution of different Gleason patterns in the eigen feature space. The K-component GMM is denoted by \(\varTheta = \{(\omega _k,\mu _k,\varSigma _k)|k=1,...,K\}\), where \(\omega _k\) is the mixing weight (\(\sum _{k=1}^{K}\omega _k = 1\)), \(\mu _k\) is the mean and \(\varSigma _k\) is the covariance matrix of the k-th mixture component. Starting with an initial mixture model, the parameters of \(\varTheta \) are estimated with Expectation-Maximization (EM) [15]. The EM algorithm is a local optimization method, and hence particularly sensitive to the initialization of the model. Instead of random initialization, we present a simple but efficient method for finding initial parameters based on our prior knowledge from pathology.
GMM Initialization: Let \(X_{H}\) be the set of all ROIs within cores of training data with the histopathology labels \(H\!\in \!\{~benign\!, GS~3+4,\! GS~4+3,\! GS~4+4~\!\}\). We first analyze the distribution of the ROIs of benign cores, \(X_{benign}\), in the eigen feature space; we observe two distinct clusters (Fig. 2) that span histopathology labels of normal and fibromuscular tissue, chronic inflammation, atrophy, and PIN. We use k-means clustering to separate the two clusters; we consider the cluster with the maximum number of “normal tissue” ROIs as the dominant benign cluster, and the second cluster as a representative for other non-cancerous tissue. Next, we use ROIs in the training dataset that correspond to the cores with GS 4+4, \(X_{GS 4+4}\), to identify the dominant cluster that represents Gleason 4 pattern. Finally, we use all other ROIs from cancerous cores that correspond to GS 3+4 and GS 4+3 to identify the centre for Gleason 3 pattern in the eigen feature space. We denote the centroid of all clusters by \(C=\{C_{benign}\), \(C_{G4}\), \(C_{G3}\), \(C_{noncancerous}\)}. To initialize the K-component GMM, we set \(K=4\) to model the four tissue patterns with mean, \(\mu _k\), for each Gaussian component equal to the centroid of each cluster. We use an equal covariance matrices for all components and set \(\varSigma _k\) to the covariance of \(X_H\). Each \(\omega _k, k = 1,...,K\) is randomly drawn from a uniform distribution between [0, 1] and normalized by \(\sum _{k=1}^{K}\omega _k\).

An illustration of the proposed GMM initialization method.
Prediction of Gleason Score: For each test core, we map the data from 20 ROIs in that core to the eigen feature space. Subsequently, we assign a label from \(\{\)benign, G3, G4, non-cancerous\(\}\) to each ROI based on its proximity to the corresponding cluster centre in the eigen feature space. To determine a GS for a test core, Y, we follow histopathology guidelines where we use the ratio of the number of ROIs labeled as benign, G3 (\(N_{G3}\)) and G4 (\(N_{G4}\)) (e.g., a core with a large number of G4 and a small number of G3 ROIs has GS 4+3):
3 Results and Discussion
We assess the overall performance of our approach using the area under the receiver operating characteristic curve (AUC). This curve depicts relative trade-offs between sensitivity and specificity where larger AUC values indicate better classification performance. Figure 3 (top) shows the target location and distribution of histopathologic outcome of biopsies in the prostate, as divided into anterior/posterior, and central/peripheral zones for base, midgland and apex. Figure 3 (bottom) shows our predictions of cancer grades using temporal US data. The distribution of cancerous cores out of all biopsies by location within the gland was 34 % (19 out of 56 biopsies) in the central region, and 24 % (25 out of 109 biopsies) in the peripheral region. Although more biopsies were performed in the peripheral zone, a higher portion of positive biopsies was observed in the central zone. In the central zone, we can differentiate between non-cancerous targets and clinically significant cancer (GS \(\ge 4+3\)) with the AUC of 0.80.
Table 1 shows the classification performance based on the inter-class AUC. To investigate the effect of the size of the tumor on our detection performance, we analyze the AUC against the greatest length of the tumor in MRI for each target biopsy ranging from 0.3 cm to 3.8 cm. We obtained AUC of 0.70 for cores with MR-tumor-size\(\ge \)2.0 cm. The results show our method has a higher performance for larger tumors.

Target location and distribution of biopsies in the test data. Light and dark gray indicate central and peripheral zones, respectively. The pie charts indicate the number of cores and their histopathology. The size of the chart is proportional to the number of biopsies (in the range from 1 to 25) and the colors dark red, light red and blue refer to cores with GS \(\ge 4+3\), GS \(\le 3+4\) and benign pathology, respectively. The top and bottom rows depict histopathology results and our grade predictions, respectively.
We also performed an analysis to determine the sensitivity of our methodology to the choice of the training data. We create 32 pairs of training and testing datasets: each new pair of datasets is identical to the original except that one benign or cancerous core is swapped between the datasets in the pair. As Table 1 shows, the average AUC of the sensitivity analysis follows our previous performance results which support the generalization of the proposed model.
Finally, we combine our cancer grading results with readings from mp-MRI. The combination takes advantage of both imaging techniques. If mp-MRI declares cancer suspicious level as low or high for a core, we use its predictions alone and declare the core as benign or aggressive cancer, respectively. On the other hand, when mp-MRI declares the suspicious level as intermediate (70 % of all cores in our data), we use predictions based on temporal US data. The combined approach leads to an AUC of 0.72 for predicting cancer grade versus either 0.65 using mp-MRI or 0.69 using temporal US data. The combined AUC is 0.83 for tumors with \(L \ge 2.0\) cm.
4 Conclusion
In this paper, in an in vivo study including 197 TRUS-guided biopsy cores, temporal US data was used to differentiating between clinically less significant prostate cancer (GS \(\le \) 3+4), aggressive prostate (GS \(\ge \) 4+3) and non-cancerous prostate tissues. Determining the aggressiveness of prostate cancer can help reduce the current high rate of over-treatment in patients with indolent cancer. We utilized a two step machine learning approach to address the challenges related to ground-truth labeling in PCa grading. First, differentiating features for detection of cancerous and non-cancerous prostate tissue were learned, and then the statistical distribution of PCa grades was modeled using a GMM. We showed that we could successfully differentiate among aggressive PCa (GS \(\ge \) 4+3), clinically less significant PCa (GS \(\le \) 3+4), and non-cancerous prostate tissues. Furthermore, combination of temporal US and mp-MRI has the potential to outperform either modality alone in detection of PCa.
Future work includes: (1) examining physical phenomena governing US time series tissue typing. Our results to-date suggest that tissue microvibration, possibly due to cardiac pulsation, and changes in tissue temperature due to acoustic energy [4] play key roles; (2) an inter-institution patient study to determine the accuracy across a wide range of patient subpopulation. By displaying the predicted grade not only for the target, but also for regions surrounding the target, we will determine if US time series can increase cancer yield.
Notes
- 1.
Surveillance, Epidemiology, and End Results (SEER) Cancer Statistics Review.
References
Azizi, S., et al.: US-based detection of PCa using automatic feature selection with deep belief networks. In: MICCAI, pp. 70–77. Springer (2015)
Bell, A.J., Sejnowski, T.J.: The independent components of natural scenes are edge filters. Vis. Res. 37(23), 3327–3338 (1997)
Correas, J.M., et al.: PCa: diagnostic performance of real-time shear-wave elastography. Radiology 275(1), 280–289 (2014)
Daoud, M., et al.: Tissue classification using US-induced variations in acoustic backscattering features. IEEE TBME 60(2), 310–320 (2013)
Epstein, J.I., et al.: Upgrading and downgrading of PCa from biopsy to radical prostatectomy: incidence and predictive factors using the modified Gleason grading system and factoring in tertiary grades. Eur. Urol. 61(5), 1019–1024 (2012)
Feleppa, E., et al.: Recent advances in ultrasonic tissue-type imaging of the prostate. In: Acoustical Imaging, pp. 331–339. Springer (2007)
Imani, F., et al.: US-based characterization of PCa using joint independent component analysis. IEEE TBME 62(7), 1796–1804 (2015)
Khojaste, A., et al.: Characterization of aggressive PCa using US RF time series. In: SPIE Med. Imaging, p. 94141A (2015)
Kuru, T.H., et al.: Critical evaluation of magnetic resonance imaging targeted, transrectal US guided transperineal fusion biopsy for detection of PCa. J. Urol. 190(4), 1380–1386 (2013)
Llobet, R., et al.: Computer-aided detection of PCa. Int. J. Med. Inform. 76(7), 547–556 (2007)
Nelson, E.D., et al.: Targeted biopsy of the prostate: the impact of color Doppler imaging and elastography on PCa detection and Gleason score. Urology 70(6), 1136–1140 (2007)
de Rooij, M., et al.: Accuracy of multiparametric MRI for PCa detection: a meta-analysis. Am. J. Roentgenol. 202(2), 343–351 (2014)
Siddiqui, M.M., et al.: Comparison of MR/US fusion-guided biopsy with US-guided biopsy for the diagnosis of PCa. JAMA 313(4), 390–397 (2015)
Vargas, H.A., et al.: Diffusion-weighted endorectal MRI at 3T for PCa: tumor detection and assessment of aggressiveness. Radiology 259(3), 775–784 (2011)
Xu, L., Jordan, M.I.: On convergence properties of the EM algorithm for Gaussian mixtures. Neural Comput. 8(1), 129–151 (1996)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this paper
Cite this paper
Azizi, S. et al. (2016). Classifying Cancer Grades Using Temporal Ultrasound for Transrectal Prostate Biopsy. In: Ourselin, S., Joskowicz, L., Sabuncu, M., Unal, G., Wells, W. (eds) Medical Image Computing and Computer-Assisted Intervention – MICCAI 2016. MICCAI 2016. Lecture Notes in Computer Science(), vol 9900. Springer, Cham. https://doi.org/10.1007/978-3-319-46720-7_76
Download citation
DOI: https://doi.org/10.1007/978-3-319-46720-7_76
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46719-1
Online ISBN: 978-3-319-46720-7
eBook Packages: Computer ScienceComputer Science (R0)