
Abstract
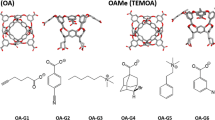
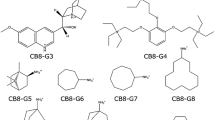
Molecular containers such as cucurbit[7]uril (CB7) and the octa-acid (OA) host are ideal simplified model test systems for optimizing and analyzing methods for computing free energies of binding intended for use with biologically relevant protein–ligand complexes. To this end, we have performed initially blind free energy calculations to determine the free energies of binding for ligands of both the CB7 and OA hosts. A subset of the selected guest molecules were those included in the SAMPL4 prediction challenge. Using expanded ensemble simulations in the dimension of coupling host–guest intermolecular interactions, we are able to show that our estimates in most cases can be demonstrated to fully converge and that the errors in our estimates are due almost entirely to the assigned force field parameters and the choice of environmental conditions used to model experiment. We confirm the convergence through the use of alternative simulation methodologies and thermodynamic pathways, analyzing sampled conformations, and directly observing changes of the free energy with respect to simulation time. Our results demonstrate the benefits of enhanced sampling of multiple local free energy minima made possible by the use of expanded ensemble molecular dynamics and may indicate the presence of significant problems with current transferable force fields for organic molecules when used for calculating binding affinities, especially in non-protein chemistries.








Similar content being viewed by others

References
Mobley DL, Dill KA (2007) Confine-and-release method: obtaining correct binding free energies in the presence of protein conformational change. J Chem Theory Comput 3:1231–1235
Gallicchio E, Lapelosa M, Levy RM (2010) Binding energy distribution analysis method (BEDAM) for estimation of protein–ligand binding affinities. J Chem Theory Comput 6:2961–2977
Boyce SE, Mobley DL, Rocklin GJ, Graves AP, Dill Ka, Shoichet BK (2009) Predicting ligand binding affinity with alchemical free energy methods in a polar model binding site. J Mol Biol 394(4):747–63
Jayachandran G, Shirts MR, Park S, Pande VS (2006) Parallelized-over-parts computation of absolute binding free energy with docking and molecular dynamics. J Chem Phys 125(8):084,901
Isaacs L (2009) Cucurbit[n]urils: from mechanism to structure and function. Chem Commun (6):619–29
Sun H, Gibb CLD, Gibb BC (2008) Calorimetric analysis of the 1:1 complexes formed between a water-soluble deep-cavity cavitand, and cyclic and acyclic carboxylic acids. Supramol Chem 20(1–2):141–147
Ong W, Kaifer AE (2004) Salt effects on the apparent stability of the cucurbit[7]uril-methyl viologen inclusion complex. J Org Chem 69(4):1383–5
Moghaddam S, Yang C, Rekharsky M, Ko YH, Kim K, Inoue Y, Gilson MK (2011) New ultrahigh affinity host–guest complexes of cucurbit[7]uril with bicyclo[2.2.2]octane and adamantane guests: thermodynamic analysis and evaluation of M2 affinity calculations. J Am Chem Soc 133(10):3570–81
Moghaddam S, Inoue Y, Gilson MK (2009) Host–guest complexes with protein–ligand-like affinities: computational analysis and design. J Am Chem Soc 131(11):4012–21
Wyman IW, Macartney DH (2008) Cucurbit[7]uril host–guest complexes with small polar organic guests in aqueous solution. Org Biomol Chem 6(10):1796–801
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) Blind prediction of the host–guest binding affinities from the SAMPL4 challenge. J Comput Aided Mol Des (in press)
Lyubartsev AP, Martsinovski AA, Shevkunov SV, Vorontsov-Velyaminov PN (1992) New approach to Monte Carlo calculation of the free energy: method of expanded ensembles. J Chem Phys 96(3):1776
Escobedo Fa, Martínez-Veracoechea FJ (2007) Optimized expanded ensembles for simulations involving molecular insertions and deletions. I. Closed systems. J Chem Phys 127(17):174103
Desgranges C, Delhommelle J (2012) Evaluation of the grand-canonical partition function using expanded Wang–Landau simulations. I. Thermodynamic properties in the bulk and at the liquid-vapor phase boundary. J Chem Phys 136(18):184107
Wang F, Landau D (2001) Efficient, multiple-range random walk algorithm to calculate the density of states. Phys Rev Lett 86(10):2050–2053
Wang K, Yang Y, Chodera JD, Shirts MR (2013) Identifying ligand binding sites and poses using GPU-accelerated Hamiltonian replica exchange molecular dynamics. J Comput Aid Mol Des 12(27):989–1007
Mobley DL (2012) Let’s get honest about sampling. J Comput Aided Mol Des 26(1):93–5
Flyvbjerg H, Petersen HG (1989) Error estimates on averages of correlated data. J Chem Phys 91(1):461
Hess B (2002) Convergence of sampling in protein simulations. Phys Rev E 65(3):1–10
Grossfield A, Zuckerman DM (2009) Quantifying uncertainty and sampling quality in biomolecular simulations. Ann Rep Comput Chem 5:23–48
Belardinelli RE, Manzi S, Pereyra VD (2008) Analysis of the convergence of the 1/t and Wang–Landau algorithms in the calculation of multidimensional integrals. Phys Rev E 78:067701
da Silva AWS, Vranken WF (2012) Acpype—antechamber python parser interface. BMC Res Notes 5:367
Wang J, Wang W, Kollman PA, Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25:247–260
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174
Jakalian A, Jack DB, Bayly CI (2002) Fast, effcient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem 23(16):1623–1641
Humphrey W, Dalke A, Schulten K (1996) VMD—visual molecular dynamics. J Mol Graph 14:33–38
Hess B, Kutzner C, Spoel DVD, Lindahl E (2008) Gromacs 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29(7):845–54
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Cao L, Isaacs L (2013) Absolute and relative binding affinity of cucurbit[7]uril towards a series of cationic guests. Supramol Chem. doi:10.1080/10610278.2013.852674
Liu DCL, Nocedal J (1989) On the limited memory method for large scale optimization. Math Program B 45(3):503–528
Martyna GJ, Tuckerman ME, Tobias DJ, Klein ML (1996) Explicit reversible integrators for extended systems dynamics. Mol Phys 87:1117–1157
Ryckaert JP, Ciccotti G, Berendsen HJ (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23(3):327–341
Andersen C (1983) RATTLE: a “velocity” version of the SHAKE algorithm for molecular dynamics calculations. J Comput Phys 52:24–34
Chodera JD, Shirts MR (2011) Replica exchange and expanded ensemble simulations as Gibbs sampling: simple improvements for enhanced mixing. J Chem Phys 135(19):194110
Paliwal H, Shirts MR (2011) A benchmark test set for alchemical free energy transformations and its use to quantify error in common free energy methods. J Chem Theory Comput 7(12):4115–4134
Chodera JD, Shirts MR (2009) A python implementation of the multistate Bennet acceptance ratio (MBAR). https://simtk.org/home/pymbar
Shirts MR, Chodera JD (2008) Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys 129(12):124105
Boresch S, Tettinger F, Leitgeb M, Karplus M (2003) Absolute binding free energies: a quantitative approach for their calculation. J Phys Chem B 107(35):9535–9551
Deng Y, Roux B (2006) Calculation of standard binding free energies: aromatic molecules in the T4 lysozyme L99A mutant. J Chem Theory Comput 2(5):1255–1273
Wang J, Deng Y, Roux B (2006) Absolute binding free energy calculations using molecular dynamics simulations with restraining potentials. Biophys J 91(8):2798–814
Hunenberger PH, McCammon JA (1999) Ewald artifacts in computer simulations of ionic solvation and ionion interaction: a continuum electrostatics study. J Chem Phys 110(4):1856
Rocklin GJ, Mobley DL, Dill Ka, Hünenberger PH (2013) Calculating the binding free energies of charged species based on explicit-solvent simulations employing lattice-sum methods: an accurate correction scheme for electrostatic finite-size effects. J Chem Phys 139(18):184103
Rogers KE, Ortiz-Sánchez JM, Baron R, Fajer M, de Oliveira CAF, McCammon JA (2013) On the role of dewetting transitions in host–guest binding free energy calculations. J Chem Theory Comput 9(1):46–53
Yang W, Bitetti-Putzer R, Karplus M (2004) Free energy simulations: use of reverse cumulative averaging to determine the equilibrated region and the time required for convergence. J Chem Phys 120(6):2618–28
Acknowledgments
The authors would like to thank the NanoSTAR Institute at the University of Virginia for an undergraduate research grant, David Mobley (UC-Irvine) for his patience and strong leadership in the SAMPL4 competition, OpenEye for sponsorship of the SAMPL4 competition, and Lyle Isaacs (University of Maryland-College Park) and Mike Gilson (UC-San Diego) for their work in preparing the host–guest systems for SAMPL4.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Monroe, J.I., Shirts, M.R. Converging free energies of binding in cucurbit[7]uril and octa-acid host–guest systems from SAMPL4 using expanded ensemble simulations. J Comput Aided Mol Des 28, 401–415 (2014). https://doi.org/10.1007/s10822-014-9716-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-014-9716-4