
Abstract
The computation of distribution coefficients between polar and apolar phases requires both an accurate characterization of transfer free energies between phases and proper accounting of ionization and protomerization. We present a protocol for accurately predicting partition coefficients between two immiscible phases, and then apply it to 53 drug-like molecules in the SAMPL5 blind prediction challenge. Our results combine implicit solvent QM calculations with classical MD simulations using the non-Boltzmann Bennett free energy estimator. The OLYP/DZP/SMD method yields predictions that have a small deviation from experiment (RMSD = 2.3 \(\log\) D units), relative to other participants in the challenge. Our free energy corrections based on QM protomer and \({\text{p}}K_{\text{a}}\) calculations increase the correlation between predicted and experimental distribution coefficients, for all methods used. Unfortunately, these corrections are overly hydrophilic, and fail to account for additional effects such as aggregation, water dragging and the presence of polar impurities in the apolar phase. We show that, although expensive, QM-NBB free energy calculations offer an accurate and robust method that is superior to standard MM and QM techniques alone.






Similar content being viewed by others
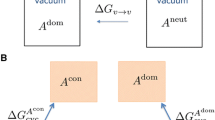
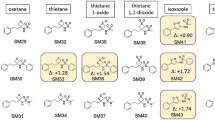
Notes
This is the version of Dunning’s DZP basis set that appears in the Psi4 quantum chemistry package [72].
References
Abraham MH, Zissimos AM, Acree WE Jr (2001) Partition of solutes from the gas phase and from water to wet and dry di-n-butyl ether: a linear free energy relationship analysis. Phys Chem Chem Phys 3:3732–3736. doi:10.1039/B104682A
Bannan CC, Burley KH, Mobley DL (2016) Blind prediction of cyclohexane-water distribution coefficients from the SAMPL5 challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9954-8
Bausch M, Selmarten D, Gostowski R, Dobrowolski P (1991) Potentiometric and spectroscopic investigations of the aqueous phase acidbase chemistry of urazoles and substituted urazoles. J Phys Org Chem 4(1):67–69. doi:10.1002/poc.610040111
Becke A (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38(6):3098–3100. doi:10.1103/PhysRevA.38.3098
Beierlein FR, Michel J, Essex JW (2011) A simple QM/MM approach for capturing polarization effects in protein-ligand binding free energy calculations. J Phys Chem B 115(17):4911–4926. doi:10.1021/jp109054j
Bennett CH (1976) Efficient estimation of free energy differences from Monte Carlo data. J Comput Phys 22:245–268
Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF (1994) Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem Phys Lett 222:529–539
Bhatnagar N, Kamath G, Chelst I, Potoff JJ (2012) Direct calculation of 1-octanolwater partition coefficients from adaptive biasing force molecular dynamics simulations. J Chem Phys 137(1):014502. doi:10.1063/1.4730040
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization and dynamics calculations. J Comput Chem 4:187–217
Brooks B, Brooks C III, Mackerell A Jr, Nilsson L, Petrella R, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner A, Feig M, Fischer S, Gao J, Hodošček M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor R, Post C, Pu J, Schaefer M, Tidor B, Venable R, Woodcock H, Wu X, Yang W, York D, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30(10, Sp. Iss. SI):1545–1614. doi:10.1002/jcc.21287
Casasnovas R, Ortega-Castro J, Frau J, Donoso J, Muoz F (2014) Theoretical pKa calculations with continuum model solvents, alternative protocols to thermodynamic cycles. Int J Quantum Chem 114(20):1350–1363. doi:10.1002/qua.24699
Cave-Ayland C, Skylaris CK, Essex JW (2015) Direct validation of the single step classical to quantum free energy perturbation. J Phys Chem B 119(3, SI):1017–1025. doi:10.1021/jp506459v
Comer J, Tam K (2007) Lipophilicity profiles: theory and measurement. Verlag Helvetica Chimica Acta, pp 275–304. doi:10.1002/9783906390437.ch17
Darden T, York D, Pedersen L (1993) Particle mesh Ewald—an N Log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Du Q, Freysz E, Shen YR (1994) Surface vibrational spectroscopic studies of hydrogen bonding and hydrophobicity. Science 264(5160):826–828. doi:10.1126/science.264.5160.826
Dunning TH (1970) Gaussian basis functions for use in molecular calculations. I. Contraction of (9s5p) atomic basis sets for the firstrow atoms. J Chem Phys 53(7):2823–2833. doi:10.1063/1.1674408
Dybeck EC, König G, Brooks BR, Shirts MR (2016) A comparison of methods to reweight from classical molecular simulations to QM/MM potentials. J Chem Theory Comput. doi:10.1021/acs.jctc.5b01188
Fan W, Tayar NE, Testa B, Kier LB (1990) Water-dragging effect: a new experimental hydration parameter related to hydrogen-bond-donor acidity. J Phys Chem 94(12):4764–4766. doi:10.1021/j100375a003
Fan W, Tsai RS, Tayar NE, Carrupt PA, Testa B (1994) Soluble-water interactions in the organic phase of a biphasic system. 2. Effects of organic phase and temperature on the “water-dragging” effect. J Phys Chem 98(1):329–333. doi:10.1021/j100052a054
Fox SJ, Pittock C, Tautermann CS, Fox T, Christ C, Malcolm NOJ, Essex JW, Skylaris CK (2013) Free energies of binding from large-scale first-principles quantum mechanical calculations: application to ligand hydration energies. J Phys Chem B 117(32):9478–9485. doi:10.1021/jp404518r
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, revision B.01. Gaussian, Inc., Wallingford
Geballe MT, Guthrie JP (2012) The SAMPL3 blind prediction challenge: transfer energy overview. J Comput Aided Mol Des 26(5):489–496. doi:10.1007/s10822-012-9568-8
Genheden S, Ryde U, Söderhjelm P (2015) Binding affinities by alchemical perturbation using QM/MM with a large QM system and polarizable MM model. J Comput Chem 36(28):2114–2124. doi:10.1002/jcc.24048
Hall HK (1957) Correlation of the base strengths of amines. J Am Chem Soc 79(20):5441–5444. doi:10.1021/ja01577a030
Handy NC, Cohen AJ (2001) Left-right correlation energy. Mol Phys 99(5):403–412. doi:10.1080/00268970010018431
Heimdal J, Ryde U (2012) Convergence of QM/MM free-energy perturbations based on molecular-mechanics or semiempirical simulations. Phys Chem Chem Phys 14:12,59212,604. doi:10.1039/c2cp41005b
Hoe WM, Cohen AJ, Handy NC (2001) Assessment of a new local exchange functional OPTX. Chem Phys Lett 341(34):319–328. doi:10.1016/S0009-2614(01)00581-4
Hoover WG (1985) Canonical dynamics—equilibrium phase-space distributions. Phys Rev A 31:1695
Hu YF, Lv WJ, Shang YZ, Liu HL, Wang HL, Suh SH (2013) Dmso transport across water/hexane interface by molecular dynamics simulation. Ind Eng Chem Res 52(19):6550–6558. doi:10.1021/ie303006d
Hudson PS, White JK, Kearns FL, Hodošček M, Boresch S, Woodcock HL (2015) Efficiently computing pathway free energies: new approaches based on chain-of-replica and Non-Boltzmann Bennett reweighting schemes. Biochim Biophys Acta Gen Subj 1850(5, SI):944–953. doi:10.1016/j.bbagen.2014.09.016
Hudson PS, Woodcock HL, Boresch S (2015) Use of nonequilibrium work methods to compute free energy differences between molecular mechanical and quantum mechanical representations of molecular systems. J Phys Chem Lett 6(23):4850–4856. doi:10.1021/acs.jpclett.5b02164
Ingram T, Storm S, Kloss L, Mehling T, Jakobtorweihen S, Smirnova I (2013) Prediction of micelle/water and liposome/water partition coefficients based on molecular dynamics simulations, cosmo-rs, and cosmomic. Langmuir 29(11):3527–3537. doi:10.1021/la305035b
Jia X, Wang M, Shao Y, König G, Brooks BR, Zhang JZH, Mei Y (2016) Calculations of solvation free energy through energy reweighting from molecular mechanics to quantum mechanics. J Chem Theory Comput 12(2):499–511. doi:10.1021/acs.jctc.5b00920
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79(2):926–935. doi:10.1063/1.445869
Klamt A (2011) The COSMO and COSMO-RS solvation models. Wiley Interdiscip Rev Comput Mol Sci 1(5):699–709. doi:10.1002/wcms.56
Klamt A (2016) Placeholder: Cosmo-rs sampl5 results. J Comput Aided Mol Des. doi:10.1007/s10822-016-9927-y
König G, Mei Y, Pickard FC, Simmonett AC, Miller BT, Herbert JM, Woodcock HL, Bernard BR, Shao Y (2016) Computation of hydration free energies using the multiple environment single system quantum mechanical/molecular mechanical method. J Chem Theory Comput 12(1):332–344. doi:10.1021/acs.jctc.5b00874
König G, Pickard FC, Huang J, Simmonett C, Tofoleanu F, Lee J, Dral PO, Prasad S, Jones M, Shao Y, Thiel W, Brooks BR (2016) Calculating distribution coefficients based on multi-scale free energy simulations an evaluation of MM and QM/MM explicit solvent simulations of water-cyclohexane transfer in the SAMPL5 challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9936-x
König G, Hudson PS, Boresch S, Woodcock HL (2014) Multiscale free energy simulations: an efficient method for connecting classical MD simulations to QM or QM/MM free energies using Non-Boltzmann Bennett Reweighting schemes. J Chem Theory Comput 10(4):1406–1419. doi:10.1021/ct401118k
König G, Pickard FC, Mei Y, Brooks BR (2014) Predicting hydration free energies with a hybrid QM/MM approach: an evaluation of implicit and explicit solvation models in SAMPL4. J Comput Aided Mol Des 28(3):245–257. doi:10.1007/s10822-014-9708-4
König G, Boresch S (2011) Non-Boltzmann sampling and bennett’s acceptance ratio method: how to profit from bending the rules. J Comput Chem 32(6):1082–1090. doi:10.1002/jcc.21687
König G, Brooks BR (2015) Correcting for the free energy costs of bond or angle constraints in molecular dynamics simulations. Biochim Biophys Acta Gen Subj 1850(5):932–943. doi:10.1016/j.bbagen.2014.09.001
Kunieda M, Nakaoka K, Liang Y, Miranda CR, Ueda A, Takahashi S, Okabe H, Matsuoka T (2010) Self-accumulation of aromatics at the oilwater interface through weak hydrogen bonding. J Am Chem Soc 132(51):18281–18286. doi:10.1021/ja107519d
Lee C, Yang W, Parr RG (1988) Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789. doi:10.1103/PhysRevB.37.785
Lee AC, Yu Yu J, Crippen GM (2008) pKa prediction of monoprotic small molecules the smarts way. J Chem Inf Model 48(10):2042–2053. doi:10.1021/ci8001815
Lin B, Pease JH (2013) A novel method for high throughput lipophilicity determination by microscale shake flask and liquid chromatography tandem mass spectrometry. Comb Chem High Throughput Screen 16(10):817–825. doi:10.2174/1386207311301010007
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) In vitro models for selection of development candidates experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23(1):3–25. doi:10.1016/S0169-409X(96)00423-1
Lipnick RL (2008) Environmental hazard assessment using lipophilicity data. Wiley-VCH Verlag GmbH, pp 339–353. doi:10.1002/9783527614998.ch19
Liptak M, Shields G (2001) Accurate pKa calculations for carboxylic acids using complete basis set and Gaussian-n models combined with CPCM continuum solvation methods. J Am Chem Soc 123(30):7314–7319. doi:10.1021/ja010534f
Marenich AV, Cramer CJ, Truhlar DG (2009) Performance of SM6, SM8, and SMD on the SAMPL1 test set for the prediction of small-molecule solvation free energies. J Phys Chem B 113(14):4538–4543
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396
McQuarrie DA (1976) Statistical mechanics. Harper and Row, New York
Miehlich B, Savin A, Stoll H, Preuss H (1989) Results obtained with the correlation energy density functionals of becke and lee, yang and parr. Chem Phys Lett 157(3):200–206. doi:10.1016/0009-2614(89)87234-3
Mikulskis P, Cioloboc D, Andrejić M, Khare S, Brorsson J, Genheden S, Mata RA, Söderhjelm P, Ryde U (2014) Free-energy perturbation and quantum mechanical study of SAMPL4 octa-acid host-guest binding energies. J Comput Aided Mol Des 28(4):375–400. doi:10.1007/s10822-014-9739-x
Mobley DL, Wymer KL, Lim NM, Guthrie JP (2014) Blind prediction of solvation free energies from the SAMPL4 challenge. J Comput Aided Mol Des 28(3):135–150. doi:10.1007/s10822-014-9718-2
Ollson MA, Söderhjelm P, Ryde U (2016) Converging ligand-binding free energies obtained with free-energy perturbations at the quantum mechanical level. J Comput Chem 37(17):1589–1600. doi:10.1002/jcc.24375
Perrin DD, Dempsey B, Serjeant EP (1981) pKa prediction for organic acids and bases. Chapman and Hall, London
Pickard IV FC, König G, Simmonett AC, Shao Y, Brooks BR (2016) An efficient protocol for obtaining accurate hydration free energies using quantum chemistry and reweighting from molecular dynamics simulations. Bioorg Med Chem. doi:10.1016/j.bmc.2016.08.031
Ribeiro RF, Marenich AV, Cramer CJ, Truhlar DG (2010) Prediction of SAMPL2 aqueous solvation free energies and tautomeric ratios using the SM8, SM8AD, and SMD solvation models. J Comput Aided Mol Des 24(4):317–333. doi:10.1007/s10822-010-9333-9
Rodinger T, Pomès R (2005) Enhancing the accuracy, the efficiency and the scope of free energy simulations. Curr Opin Struct Biol 15:164–170
Rustenburg AS, Dancer J, Lin B, Feng JA, Ortwine DF, Mobley DL, Chodera JD (2016) Measuring experimental cyclohexane-water distribution coefficients for the SAMPL5 challenge. J Comput Aided Mol Des. doi:10.1007/s10822-016-9971-7
Ryde U, Söderhjelm P (2016) Ligand-binding affinity estimates supported by quantum-mechanical methods. Chem Rev 116(9):5520–5566. doi:10.1021/acs.chemrev.5b00630
Sampson C, Fox T, Tautermann CS, Woods C, Skylaris CK (2015) A “Stepping Stone” approach for obtaining quantum free energies of hydration. J Phys Chem B 119(23):7030–7040. doi:10.1021/acs.jpcb.5b01625
Shirts MR, Chodera JD (2008) Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys 129(12):124105. doi:10.1063/1.2978177
Skillman AG, Geballe MT, Nicholls A (2010) SAMPL2 challenge: prediction of solvation energies and tautomer ratios. J Comput Aided Mol Des 24(4):257–258. doi:10.1007/s10822-010-9358-0
Speight JG (2005) Lange’s handbook of chemistry, 16th edn. McGraw-Hill Education, New York
Sugita Y, Kitao A, Okamoto Y (2000) Multidimensional replica-exchange method for free-energy calculations. J Chem Phys 113:6042–6050
Sugita Y, Okamoto Y (1999) Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett 314:141–151
Tissandier MD, Cowen KA, Feng WY, Gundlach E, Cohen MH, Earhart AD, Coe JV, Thomas R, Tuttle J (1998) The proton’s absolute aqueous enthalpy and gibbs free energy of solvation from cluster-ion solvation data. J Phys Chem A 102(40):7787–7794. doi:10.1021/jp982638r
Tofoleanu F, Brooks BR, Buchete NV (2015) Modulation of Alzheimers a protofilament-membrane interactions by lipid headgroups. ACS Chem Neurosci 6(3):446–455. doi:10.1021/cn500277f
Torrie GM, Valleau JP (1977) Nonphysical sampling distributions in monte carlo free-energy estimation: umbrella sampling. J Comput Phys 23:187
Turney JM, Simmonett AC, Parrish RM, Hohenstein EG, Evangelista FA, Fermann JT, Mintz BJ, Burns LA, Wilke JJ, Abrams ML, Russ NJ, Leininger ML, Janssen CL, Seidl ET, Allen WD, Schaefer HF, King RA, Valeev EF, Sherrill CD, Crawford TD (2012) Psi4: an open-source ab initio electronic structure program. Wiley Interdiscip Rev Comput Mol Sci 2(4):556–565. doi:10.1002/wcms.93
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD Jr (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31(4):671–690. doi:10.1002/jcc.21367
Verdolino V, Cammi R, Munk BH, Schlegel HB (2008) Calculation of pka values of nucleobases and the guanine oxidation products guanidinohydantoin and spiroiminodihydantoin using density functional theory and a polarizable continuum model. J Phys Chem B 112(51):16860–16873. doi:10.1021/jp8068877
Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, Romero DL, Masse C, Knight JL, Steinbrecher T, Beuming T, Damm W, Harder E, Sherman W, Brewer M, Wester R, Murcko M, Frye L, Farid R, Lin T, Mobley DL, Jorgensen WL, Berne BJ, Friesner RA, Abel R (2015) Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc 137(7):2695–2703. doi:10.1021/ja512751q
Zacharias M, Straatsma TP, McCammon JA (1994) Separation-shifted scaling, a new scaling method for Lennard-Jones interactions in thermodynamic integration. J Chem Phys 100:9025–9031
Zhang S, Baker J, Pulay P (2010) A reliable and efficient first principles-based method for predicting pKa values. 1. Methodology. J Phys Chem A 114(1):425–431. doi:10.1021/jp9067069
Zhao Y, Truhlar DG (2007) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor Chem Acc 120:215–241
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41:157–167
Zwanzig RW (1954) High-temperature equation of state by a perturbation method. 1. Nonpolar gases. J Chem Phys 22:1420–1426
Acknowledgments
This work was supported by the intramural research program of the National Heart, Lung and Blood Institute of the National Institutes of Health and utilized the high-performance computational capabilities of the LoBoS and Biowulf Linux clusters at the National Institutes of Health. (http://www.lobos.nih.gov and http://biowulf.nih.gov)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pickard, F.C., König, G., Tofoleanu, F. et al. Blind prediction of distribution in the SAMPL5 challenge with QM based protomer and pK a corrections. J Comput Aided Mol Des 30, 1087–1100 (2016). https://doi.org/10.1007/s10822-016-9955-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-016-9955-7