
Abstract
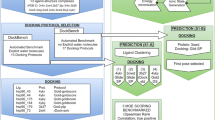
Pose prediction and virtual screening performance of a molecular docking method depend on the choice of protein structures used for docking. Multiple structures for a target protein are often used to take into account the receptor flexibility and problems associated with a single receptor structure. However, the use of multiple receptor structures is computationally expensive when docking a large library of small molecules. Here, we propose a new cross-docking pipeline suitable to dock a large library of molecules while taking advantage of multiple target protein structures. Our method involves the selection of a suitable receptor for each ligand in a screening library utilizing ligand 3D shape similarity with crystallographic ligands. We have prospectively evaluated our method in D3R Grand Challenge 2 and demonstrated that our cross-docking pipeline can achieve similar or better performance than using either single or multiple-receptor structures. Moreover, our method displayed not only decent pose prediction performance but also better virtual screening performance over several other methods.
Graphical Abstract








Similar content being viewed by others

References
Yuriev E, Agostino M, Ramsland PA (2011) Challenges and advances in computational docking: 2009 in review. J Mol Recognit 24:149–164
Yuriev E, Holien J, Ramsland PA (2015) Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J Mol Recognit 28:581–604
Yuriev E, Ramsland PA (2013) Latest developments in molecular docking: 2010–2011 in review. J Mol Recognit 26:215–239
Kutchukian PS, Shakhnovich EI (2010) De novo design: balancing novelty and confined chemical space. Expert Opin Drug Discov 5:789–812
Loving K, Alberts I, Sherman W (2010) Computational approaches for fragment-based and de novo design. Curr Top Med Chem 10:14–32
Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503:295–299
Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci USA 108:13118–13123
Buch I, Giorgino T, De Fabritiis G (2011) Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc Natl Acad Sci USA 108:10184–10189
Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD (2015) Molecular docking and structure-based drug design strategies. Molecules 20:13384–13421
Jiang X, Kumar A, Liu T, Zhang KY, Yang Q (2016) A novel scaffold for developing specific or broad-spectrum chitinase inhibitors. J Chem Inf Model 56:2413–2420
Matsuoka M, Kumar A, Muddassar M, Matsuyama A, Yoshida M, Zhang KY (2017) Discovery of fungal denitrification inhibitors by targeting copper nitrite reductase from Fusarium oxysporum. J Chem Inf Model 57:203–213
Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY (2014) Identification of sumoylation inhibitors targeting a predicted pocket in Ubc9. J Chem Inf Model 54:2784–2793
Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KY (2013) Identification of sumoylation activating enzyme 1 inhibitors by structure-based virtual screening. J Chem Inf Model 53:809–820
Kumar A, Ito A, Takemoto M, Yoshida M, Zhang KYJ (2014) Identification of 1,2,5-oxadiazoles as a new class of SENP2 inhibitors using structure based virtual screening. J Chem Inf Model 54:870–880
Yang X, Li F, Konze KD, Meslamani J, Ma A, Brown PJ, Zhou MM, Arrowsmith CH, Kaniskan HU, Vedadi M, Jin J (2016) Structure-activity relationship studies for enhancer of zeste homologue 2 (EZH2) and enhancer of zeste homologue 1 (EZH1) inhibitors. J Med Chem 59:7617–7633
Cao X, Sun Z, Cao Y, Wang R, Cai T, Chu W, Hu W, Yang Y (2014) Design, synthesis, and structure-activity relationship studies of novel fused heterocycles-linked triazoles with good activity and water solubility. J Med Chem 57:3687–3706
Pagadala NS, Syed K, Tuszynski J (2017) Software for molecular docking: a review. Biophys Rev 9:91–102
Sotriffer C (2015) Protein-Ligand Docking: From Basic Principles to Advanced Applications. In: In Silico Drug Discovery and Design. CRC Press, Boca Raton, pp 155–188
Wong CF (2015) Flexible receptor docking for drug discovery. Expert Opin Drug Discov 10:1189–1200
Grinter SZ, Zou X (2014) Challenges, applications, and recent advances of protein-ligand docking in structure-based drug design. Molecules 19:10150–10176
Chen YC (2015) Beware of docking! Trends Pharmacol Sci 36:78–95
Antunes DA, Devaurs D, Kavraki LE (2015) Understanding the challenges of protein flexibility in drug design. Expert Opin Drug Discov 10:1301–1313
Chandrika BR, Subramanian J, Sharma SD (2009) Managing protein flexibility in docking and its applications. Drug Discov Today 14:394–400
Bottegoni G, Rocchia W, Rueda M, Abagyan R, Cavalli A (2011) Systematic exploitation of multiple receptor conformations for virtual ligand screening. PLoS ONE 6:e18845
Totrov M, Abagyan R (2008) Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol 18:178–184
Kumar A, Zhang KYJ (2012) Computational fragment-based screening using RosettaLigand: the SAMPL3 challenge. J Comput-Aided Mol Des 26:603–616
Rueda M, Bottegoni G, Abagyan R (2010) Recipes for the selection of experimental protein conformations for virtual screening. J Chem Inf Model 50:186–193
Li Y, Kim DJ, Ma W, Lubet RA, Bode AM, Dong Z (2011) Discovery of novel checkpoint kinase 1 inhibitors by virtual screening based on multiple crystal structures. J Chem Inf Model 51:2904–2914
Xu M, Lill MA (2012) Utilizing experimental data for reducing ensemble size in flexible-protein docking. J Chem Inf Model 52:187–198
Huang Z, Wong CF (2016) Inexpensive method for selecting receptor structures for virtual screening. J Chem Inf Model 56:21–34
Ye Z, Baumgartner MP, Wingert BM, Camacho CJ (2016) Optimal strategies for virtual screening of induced-fit and flexible target in the 2015 D3R grand challenge. J Comput Aided Mol Des 30:695–706
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242
Krissinel E, Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268
Hawkins PC, Nicholls A (2012) Conformer generation with OMEGA: learning from the data set and the analysis of failures. J Chem Inf Model 52:2919–2936
Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 50:572–584
Hawkins PC, Skillman AG, Nicholls A (2007) Comparison of shape-matching and docking as virtual screening tools. J Med Chem 50:74–82
ROCS 3.2.0.4: OpenEye Scientific Software, Santa Fe, NM
Rogers DJ, Tanimoto TT (1960) A computer program for classifying plants. Science 132:1115–1118
Schrödinger (2015) Release 2015-3: LigPrep, version 3.5, Schrödinger. LLC, New York, NY
Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM (2005) Integrated modeling program, applied chemical theory (IMPACT). J Comput Chem 26:1752–1780
Schrödinger (2015) Release 2015-3: Maestro, version 10.3, Schrödinger. LLC, New York, NY
Glide (2011) version 5.7, Schrödinger. LLC, New York
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47:1750–1759
Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M (2007) Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J Comput-Aided Mol Des 21:681–691
Greenwood JR, Calkins D, Sullivan AP, Shelley JC (2010) Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J Comput-Aided Mol Des 24:591–604
Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) A critical assessment of docking programs and scoring functions. J Med Chem 49:5912–5931
Plewczynski D, Lazniewski M, Augustyniak R, Ginalski K (2011) Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J Comput Chem 32:742–755
Kumar A, Zhang KY (2016) A pose prediction approach based on ligand 3D shape similarity. J Comput Aided Mol Des 30:457–469
Gathiaka S, Liu S, Chiu M, Yang H, Stuckey JA, Kang YN, Delproposto J, Kubish G, Dunbar JB, Carlson HA, Burley SK, Walters WP, Amaro RE, Feher VA, Gilson MK (2016) D3R grand challenge 2015: evaluation of protein–ligand pose and affinity predictions. J Comput-Aided Mol Des 30:651–668
Bender A, Mussa HY, Glen RC, Reiling S (2004) Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J Chem Inf Comput Sci 44:1708–1718
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Velec HF, Gohlke H, Klebe G (2005) DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J Med Chem 48:6296–6303
Acknowledgements
We acknowledge RIKEN ACCC for the supercomputing resources at the Hokusai GreatWave supercomputer used in this study. We acknowledge RIKEN Pioneering Project in Dynamic Structural Biology for funding. We thank members of our lab for help and discussions.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary material
Below is the link to the electronic Supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, A., Zhang, K.Y.J. A cross docking pipeline for improving pose prediction and virtual screening performance. J Comput Aided Mol Des 32, 163–173 (2018). https://doi.org/10.1007/s10822-017-0048-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-017-0048-z