
Abstract
In biological research, alignment of protein sequences by computer is often needed to find similarities between them. Although results can be computed in a reasonable time for alignment of two sequences, it is still very central processing unit (CPU) time-consuming when solving massive sequences alignment problems such as protein database search. In this paper, an optimized protein database search method is presented and tested with Swiss-Prot database on graphic processing unit (GPU) devices, and further, the power of CPU multi-threaded computing is also involved to realize a GPU-based heterogeneous parallelism. In our proposed method, a hybrid alignment approach is implemented by combining Smith–Waterman local alignment algorithm with Needleman–Wunsch global alignment algorithm, and parallel database search is realized with compute unified device architecture (CUDA) parallel computing framework. In the experiment, the algorithm is tested on a lower-end and a higher-end personal computers equipped with GeForce GTX 750 Ti and GeForce GTX 1070 graphics cards, respectively. The results show that the parallel method proposed in this paper can achieve a speedup up to 138.86 times over the serial counterpart, improving efficiency and convenience of protein database search significantly.
Similar content being viewed by others

1 Introduction
Searching from protein database, a lot of protein sequence information is stored, which is an essential approach in the research on bioinformatics [1]. Through the approach of protein database search that aligning a query protein sequence to sequences in database one after another, researchers could get relevant information of the query sequence from the search results. The essential part of protein database search is sequence alignment algorithm. Sequence alignment algorithms can be classified into two types: global alignment and local alignment. The Needleman–Wunsch (NW) algorithm is a global alignment method which uses dynamic programming (DP) strategy to find the optimal alignment between two sequences [2]. The Smith–Waterman (SW) algorithm is a local alignment method and it is also based on DP strategy. Unlike the NW algorithm, the SW algorithm allows finding the optimal alignment of two fragments of the studied sequences [3]. Although more computational accurate compared to prevalent heuristic alignment algorithms such as Blast [4] and Fasta [5], the NW and the SW algorithms are still restricted because exponential computation complexity requires much more run time by these methods, especially considering of the rapid expansion of sequence databases. With the development of computer science and the enhancement of hardware performance, parallel computing has been widely used as a high performance computing tool in the scientific calculation domain. There are many researches dedicated to improve the computational efficiency of compute-intensive algorithms such as DP by introducing the parallel computing technologies, and experiments of these researches show that parallel computing can reduce computing time and ensure accurate results [6].
In recent years, further improvement of CPU clock rate is limited by power consumption, heat dissipation and other issues, while the many-core computing technology represented by GPU is becoming a more powerful tool, and researches based on GPU parallel computing have received much more attentions [7]. The GPU architecture is characterized by large number of processing units, great memory bandwidth and relatively less costs. For example, the GeForce GTX 1070 GPU [8], which is based on the Pascal architecture of NVIDIA, has 1920 compute units and 256 GB/sec memory bandwidth that could guarantee more powerful parallel computing ability. Therefore, any computer that equipped with a graphics card could be operated as a cost-effective parallel computing system. Meanwhile, in the aspect of software development environments, the skill barriers of parallel programming are being reduced with the improvement of GPU parallel programming frameworks such as CUDA [9] and OpenCL [10], which further promotes the popularity of general-purpose computation on GPUs (GPGPU). Recently, the application of GPGPU technology has been extended to the field of life sciences, and previous researches have demonstrated that GPU parallel can effectively accelerate the speed of sequence database search based on the DP algorithm and the speedup is better than the CPU parallel approach obviously. However, the previous works of protein database search based on GPUs are implemented using merely one kind of alignment algorithm, local or global. They are unable to align sequences on both local and global levels; as a result, it is difficult to fully understand the similarities between sequences.
In this paper, a GPU-based parallel algorithm for hybrid search of protein database is designed and implemented with CUDA parallel computing technology. Our GPU algorithm also uses CPU as an assisted search engine to maximize the computing speed. The hybrid search approach is introduced by combining the Smith–Waterman and the Needleman–Wunsch two alignment algorithms into one search process to achieve alignments both on local and global levels. In the implement stage, ELL storage format combined with partitioned database is used to optimize the storage structure, and high-speed shared memory on GPU cores is used to improve access speed further. In our experiment, the Swiss-Prot protein database was used as a test, and the new parallel algorithm was tested on two desktop computers equipped with a GTX 1070 and a GTX 750 Ti graphics cards, respectively. The results show that the new method proposed in this paper can achieve a speedup up over 138 times and save over 95% the run time compared to the serial method.
The rest of this paper is organized as follows: First, we introduce the outline of GPU parallel computing, the protein database search process, the alignment algorithms, the sense to do the hybrid alignment and the related works in Sect. 2. Second, the design and implementation of our GPU algorithm are discussed in Sect. 3. Third, our experiment method and performance evaluation of result are described in Sect. 4. Finally, we give the conclusion of this paper and propose the future work.
2 Background
2.1 GPU parallel computing
GPUs are different from the traditional CPUs in the aspect of hardware architectures. CPUs are designed for executing serial code. Branch prediction unit, multi-level cache and other hardware structures enable the efficient serial execution of CPUs. GPUs are designed for working in compute-intensive and highly data parallel scenarios. Because of the large number of compute units equipped by GPUs, they are suitable for parallel computing in a single instruction, multiple data (SIMD) mode. The streaming processor (SP) is the basic processing unit of GPUs; instructions and data are executed and processed within it. A streaming multiprocessor (SM) is composed of multiple SPs, shared memory, registers and other hardware resources, and there are several SMs in a GPU device generally. Since the GPUs are originally designed to weaken the branch prediction, multi-level caches and other hardware facilities, in turn, increase more SP units, and they are more competent in parallel computing compared to CPUs [11].
The programming framework that we use to implement the parallel algorithm on GPU devices is CUDA. CUDA is an open programming framework for GPU parallel computing developed and supported by NVIDIA. As an extension of the C/C++ language, we can use the standard C/C++ to develop CUDA programs. The code of CUDA consists of the host code which is executed by CPU and the device code which is executed by GPU. The device code can be called by kernel method from the host code to start multi-threaded computing tasks on GPUs.
2.2 Database search process
Protein database search is one process that can align an unknown protein sequence, which we call query sequence, with known sequences in database one after another. When comparing two sequences, we need an algorithm combined with some scoring rules to compute the final alignment score which can finally indicate the similarity level. After an overall database searching, we can find out the similar sequences in the database according to the alignment scores.
The hybrid algorithm proposed in this paper is based on the integration of the Smith–Waterman local alignment algorithm and the Needleman–Wunsch global alignment algorithm. BLOSUM62 is used as the scoring matrix [12]. We use affine gap penalty scheme in the alignment process; that is to say, the cost of the first gap is higher when inserting continuous gaps. This setting reflects the fact that opening a gap has lower possibility than extending preceding gaps in biology, making the algorithm more conforming to the biological evolution process [13].
2.3 Smith–Waterman local alignment algorithm
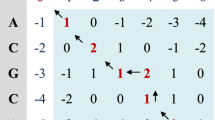
Smith–Waterman is the most widely used pairwise biological sequences local alignment algorithm based on the dynamic programming strategy. The first step of the algorithm is to construct an assisted DP matrix. As is shown in Fig. 1, query sequence “GAC” and target sequence “CGTC” construct a DP matrix whose size is \(4\times 5\). The cells of the matrices first row and column are initialized to zero. Each element of the matrix \(H_{i,j}\) is computed by a recursive way, and the recursive equation is listed below (see Eq. 1) [14].

Assisted dynamic programming matrix
In Eq. (1), \(E_{i,j}\) and \(F_{i,j}\) are two assisted matrices when computing \(H_{i,j}\), recording cost of gaps in query sequence and target sequence, respectively. Parameters \(g_o\) and \(g_e\) are gap opening cost and gap extending cost in the affine gap penalty scheme. Function \(S(q_i, t_j)\) can obtain the alignment score between the query sequence residue \(q_i\) and the target sequence residue \(t_j\) by referring scoring matrix. Algorithm 1 presents the pseudo-code of the SW algorithm.
After all the cells of the DP matrix are calculated, we can get the optimal alignment score according to the maximum value in the matrix. The optimal alignment subsequence can also be obtained by backtracking along the maximum value. In the sequence database search process, the main objective is to compute the alignment scores between query sequence and database sequences to find out similar ones, so the backtracking step for obtaining the optimal subsequence can be ignored. As needed, it can be exclusively done against similar sequences which are found in the previous database search procedure.

2.4 Needleman–Wunsch global alignment algorithm
Needleman–Wunsch is another alignment algorithm for pairwise global alignments which is also based on the dynamic programming strategy. The optimal global alignment between two sequences can be computed through this algorithm. The elements of the DP matrix \(H_{i,j}\) are computed by the recursive equation Eq. (2), and the first row and the first column of the matrix are initialized through Eq. (3). The symbols in Eqs. (2) and (3) are the same as those in Eq. (1) by definition.
The pseudo-code of the NW algorithm is given in Algorithm 2. Compared with the SW algorithm, the NW algorithm differs in the following aspects. First, the comparison with zero is not needed when computing the element \(H_{i,j}\) of the DP matrix in the NW algorithm; that is to say, the value of \(H_{i,j}\) may be negative. Second, the optimal alignment score generated by the SW algorithm is the value of the maximum element in the DP matrix, and for the NW algorithm, it is the value of the right bottom element in the DP matrix. Therefore, it is no need to calculate and save the temporary optimal alignment score in each iteration of the DP matrix computing procedure.

2.5 The hybrid alignment approach
We present a GPU-accelerated algorithm for biological sequence database search by combining the Needleman–Wunsch algorithm and the Smith–Waterman algorithm together in order to perform global and local alignments jointly. As we know, the local alignment algorithm is a regular way used in sequence database search partly because the huge difference in length between the query sequence and most of the sequences in database; therefore, the local algorithm could be more competent under the given condition. The motivation of using a hybrid alignment approach is based on the following reasons. First, for the database sequences those are similar to the query sequence in length, it is reasonable to study the similarities in a global perspective from the results of global alignment; thus, searching with a combination of local and global algorithms could save some time than doing with each of the algorithms independently; second, Polyanovsky et al. [15] demonstrated that in some conditions the global alignment method is more accurate than the local alignment method. In their work, when aligning two sequences of similar length, they revealed the global algorithm is more stable at longer evolutionary distances and larger non-homologous parts than the local algorithm on the condition that the non-homologous parts of each sequence are positioned symmetrically.
2.6 Related work
In recent years, GPUs have become an important accelerator of arithmetic computations for researchers and engineers who face large computational problems frequently. In the field of bioinformatics, GPU parallel computing technology has also been widely used as an effective tool of accelerating calculations due to the computing power of GPUs. In order to improve the speed of biological sequence alignments, following works have adopted the parallel computing technology to accelerate sequence alignment processes and achieved ideal results.
Liu et al. [16] present a parallel Smith–Waterman algorithm on GPU platform and adopt a double affine gap penalty scheme. They use a query sequence with 16384 residues and a database with 983 protein sequences in the search experiment. Compared with performance of the serial algorithm on a Pentium D 840 CPU, they report speedups of up to 4.6 times on a GeForce 7800 GTX GPU device.
Manavski and Valle [17] design and implement a GPU version of Smith–Waterman algorithm using CUDA framework. Their experiments adopt the BLOSUM50 scoring matrix and the Swiss-Prot database with 250296 protein sequences in it. They obtain performance up to 1.8 and 3.5 giga cell updates per second (GCUPS) when using one and two GeForce 8800 video cards, respectively. They demonstrate that GPUs can effectively improve the speed of biological sequence alignments.
Khajeh-Saeed et al. [18] present a performance evaluation of parallel Smith–Waterman algorithm. They use the parallel SW algorithm provided in the SSCA#1 benchmark suite and experiment on three different types of GPU devices. Their experiments indicate speedups up to 45 times when using one GPU device, and linear speedups can be obtained when using two or four GPU devices.
Blazewicz et al. [19] propose a protein alignment algorithm with a backtracking routine on GPU platform. They evaluate and compare performances of the parallel algorithm in single GPU and multi-GPU cases. The experiment results achieve 6.3 GCUPS with the affine gap penalty scheme in the case of one GPU used, and linear speedup lift of the parallel algorithm is also demonstrated when using two to four GPUs.
Siriwardena and Ranasinghe [20] implement a GPU-based parallel Needleman–Wunsch global alignment algorithm with CUDA programming framework. Testing on a GeForce 8800 GT video card, they report speedups up to 4.3 times over the serial performance on a 2.4 GHz Intel Quad Core processor.
The previous works have introduced the GPU parallel computing technology into the processes of biological sequence alignment for improving the speed of searching sequence databases and demonstrated that GPU parallelism could effectively accelerate the searching speed. However, the previous works of protein database search based on GPUs are implemented using merely one kind of alignment algorithm, local or global; that is to say, only one kind of result of the two can be obtained in one search operation. Thus, they are unable to align sequences on both local and global levels; as a result, it is difficult to fully understand the similarity between pairwise sequences especially when their lengths are close. Further, with the upgrading of GPU hardwares, it is necessary to test parallel alignment algorithms on new hardware devices and make targeted optimizations.
3 Design and implement of GPU algorithm
3.1 Parallel algorithm design
The major component of our sequence database search method is the SW–NW hybrid alignment algorithm. In this section, we first give the general flow of the database search algorithm, and then, the hybrid mode of the SW and the NW algorithms is discussed. Figure 2 illustrates the design of our parallel database search algorithm accelerated by GPUs. In this design, the complete database search algorithm is divided into the host code and the device code. The host code which is executed by CPU handles the algorithm control flow and the memory management. The most time-consuming part of the database search algorithm is the SW–NW hybrid alignment procedure, which is part of the device code and could be accelerated in parallel by GPUs.
By comparing Algorithms 1 and 2, we can find great similarities between the SW and the NW algorithms. First, in order to iterate each element in the DP matrix, both the SW and the NW algorithms contain a nested loop structure which is made of an inner loop and an outer loop. According to this feature, we implement the iteration function of the hybrid algorithm by merging together the two nested loop structures which, respectively, belong to the SW and the NW algorithms. Second, in the hybrid algorithm, the results of referring the scoring matrix could be shared by the SW and the NW computations; therefore, the function of referring the scoring matrix needs to be called only once in each iteration to reduce the run time of function calls. Third, at the code level, some computation variables could be shared between the two algorithms to save the GPU register space since they have the similar computation process.

Flowchart of our database search algorithm

Mapping strategies between GPU threads and database sequences
3.2 Mapping strategies between GPU threads and database sequences
In this paper, the proposed algorithm adopts a SIMD parallel manner. With the SIMD manner, the GPU threads execute the same sequence alignment procedure, but each thread aligns a separated database sequence. This parallel manner is similar to the one adopted in [17] and [19]. Another classic manner to parallel alignment algorithms is to compute along anti-diagonals of the DP matrix [16, 18, 20] which is based on the idea that computations of cells on the anti-diagonals are independent and can be processed in parallel. Our algorithm is designed with the SIMD manner mainly because the anti-diagonal manner is more suitable for the pairwise alignment with long sequences. Depending on the device model, usually the GPU could run hundreds to thousands of concurrent threads; however, the number of protein sequences in the database can be hundreds of thousands. Therefore, a GPU thread has to be dispatched to perform multiple sequence alignment tasks when the parallel database search is conducted. As shown in Fig. 3, we suppose that n is the number of GPU threads that we use in parallel computing and take the threads t1 and t2 as example. In this context, the sequences s(1), s(n), s(2n), etc. in the database are dispatched to the thread t1 to perform alignment and the sequences s(2), \(s(n+1)\), \(s(2n+1)\), etc. in the database are dispatched to the thread t2 to perform alignment. The computations of the threads t1 and t2 are independent and could be performed in parallel.
3.3 Database partition and ELL storage optimization
The protein sequence database Swiss-Prot, which is maintained by EBI (European Bioinformatics Institute), was applied in our experiment. In the January 2015 release of the Swiss-Prot database, there are approximately 550000 sequences, among which the shortest sequence includes 2 residues and the longest one includes 35213 residues. In order to improve the efficiency of parallel computing, we optimize the database storage structure with the data partitioning and the ELL storage format. The ELL storage format is derived from the sparse matrix toolkit ELLPACK, and its main idea is to use matrix transposition to solve the problem of un-coalesced memory access and control branching [21].
Figure 4 shows the storage optimization process of the sequence database. Sequences in the database are sorted by length first (see Fig. 4a), and then, the database storage matrix is transposed based on the ELL storage format for coalesced memory access of GPU threads (see Fig. 4b). If the transposition is not conducted, the memory has to be accessed by the threads for many times to fetch the data, because the residues of two adjacent sequences in the same position are not neighboring in the memory considering the matrix is row major. But matrix transposition successfully solved this problem, and as a result, the memory access is coalesced.
Varying sequence lengths lead to the control branching problem which makes threads have to check that if the current aligned sequence is ended. This results in performance reduction and waste of storage spaces. In order to solve the problem, we split the storage matrix of the sequence database into several partitions, and the sequences with similar length are stored in the same partition so that the storage spaces are saved effectively (see Fig. 4c). For each partition, unused spaces of the storage matrix are padded based on the idea of ELL padding (see Fig. 4d). This makes the sequence lengths are consistent in one partition and avoids the control branching problem. With the introduction of all the above methods, the speed of memory access and computation on GPUs could be increased further.

Database partition and ELL storage structure optimization
3.4 High-speed GPU cache optimization
There are many memory access operations in the device code which is executed on GPUs, leading to poor computation efficiency because of the long memory access cycle. To alleviate this problem, the high-speed shared memory on GPU cores is used to optimize our parallel algorithm. Figure 5 illustrates that the shared memory is an on-chip storage facility in the GPU architecture. The capacity of shared memory is only 48KB on current GPU architectures such as Kepler and Maxwell, but its access speed is about 10 times faster than the speed of global memory, and the data in it can be shared within all the threads belonging to the same one thread block [22]. Based on this, the use of shared memory should contribute to the improvement of the computing speed of our parallel algorithm.

GPU storage architecture
In the GPU parallel algorithm, the scoring matrix is unique, and all threads apply the same matrix to execute alignment operations. Moreover, the scoring matrix has limited and fixed storage size, so we load it into the shared memory before the beginning of alignment operations, implementing faster access of the scoring matrix by threads in computation process. Meanwhile, a lot of time is spent on reading and writing temporary data between iterations, so saving these data into the shared memory could also improve the computing efficiency.
3.5 Coupling CPU multi-threaded computing
As shown in Fig. 2, we also use CPU as an assisted computing engine besides GPU computations in our algorithm in order to guarantee better alignment speed. The CPU computation part adopts POSIX threads (Pthreads) multi-threaded operations to align different database sequences with different CPU threads in parallel, and it is executed concurrently with the GPU computations as well. Considering the sequences with more residues have lower density than those with less residues in the database, CPU is assigned to process the database sequences with length exceeding a threshold to avoid the control branching problem of GPU, and the threshold is set to 1500 in our experiment. The mapping strategy between CPU threads and database sequences is similar to the GPU’s which makes multiple CPU threads to map sequences one after another for having each thread hold the same number of sequences as much as possible.
3.6 Algorithm implement
According to the above analysis, we implement the parallel algorithm of protein database search based on hybrid alignment. In the following section, we present the experiments of the parallel algorithm to verify the accuracy and the computational efficiency. In order to more clearly demonstrate the implementation details of this method, Listing 1 gives part of the pseudo-code of carrying out the SW–NW hybrid alignment in the GPU kernel function.

4 Experiment and results
4.1 Experiment method
The algorithms evaluated in the experiment are listed in Table 1. The proposed GPU parallel algorithms SwNw_GPU and SwNw_MIX are implemented using the CUDA toolkit and the C++ programming language; moreover, the POSIX threads for Windows toolkit are employed to provide Pthreads operations for CPU multi-threaded computing module in SwNw_MIX. To evaluate the performance, we have tested our parallel algorithms on two mainstream personal computers. All the computers use Windows 10 64-bit operating system and the detailed hardware specifications are given in Table 2. In order to make a better evaluation, we have also implemented a serial CPU algorithm that integrates both the SW and the NW algorithms to compare results with the parallel algorithm in the performance test.
We use the January 2015 release of the Swiss-Prot database in our experiment. The database is about 0.3 GB in size and can be copied from the hosts to the GPU devices entirely during operations. The time it takes to complete the database transfer is far less than 0.05 second in our test and it could be ignored; furthermore, the performance could benefit from the fact that the database in GPU memory can be reused multiple times with different query sequences. We randomly select four protein sequences as the query sequences. Table 3 presents the accession number and number of residues of each query sequence. In the experiment, the gap open penalty \(g_o\) and the gap extent penalty \(g_e\) are set to \(-10\) and \(-2\), respectively. For each one of the query sequences, we search ten times in every PC platform, and the average value of execution time is used for comparisons. In addition to run time comparisons, we also use the other two evaluation criterions, GCUPS and parallel speedup. The GCUPS is a frequently used criterion to evaluate the performance of biological sequence alignment algorithms. The computation of GCUPS is defined by Eq. (4). Parameters m and n are the length of two sequences to be aligned, separately; t is the execution time of alignment. The speedup is used for measuring the performance and the effectiveness of parallel algorithms, and it is defined by Eq. (5).
4.2 Swiss-Prot database search experiment
Figure 6 gives the search results of the Swiss-Prot database using our GPU-based hybrid alignment algorithm. The search result of hybrid alignment algorithm is composed of two parts, the NW scores and the SW scores; thus, from one hybrid search operation, we could examine the query sequence in both global and local perspectives. As a global alignment algorithm, the NW is generally used to align sequences with similar lengths. Therefore, the hybrid alignment approach is set to work only when the query sequence and the database sequence are similar in length. A threshold value for the hybrid alignment is set to be 20% in the experiment, which means the hybrid alignment will only apply to the database sequences length of between 80 and 120% of query sequence length.

Swiss-Prot database searching results of our GPU hybrid alignment algorithm; the horizontal axis represents sequences in the database which are sorted by length. a Searching results of sequence WP_011236883. b Searching results of sequence P81288. c Searching results of sequence WP_002483038. d Searching results of sequence ANI98813
4.3 Performance evaluation
The run time comparison result of the parallel and the serial algorithms are shown in Fig. 7. The four sub-figures from left to right show the result corresponding to each query sequence. We can see from the figure that our parallel algorithms SwNw_GPU and SwNw_MIX exceed the serial algorithm SwNw_CPU in computing efficiency apparently. With the help of CPU computing, the GPU+CPU heterogeneous parallel algorithm SwNw_MIX outperforms the pure GPU algorithm SwNw_GPU for all the query sequences with the highest performance promotion of 17% (49%) on PC1 (PC2). Compared to the run time of the serial algorithm SwNw_CPU, the parallel algorithm SwNw_MIX could save 95.00% (95.68%), 94.59% (95.40%), 93.29% (95.31%), 93.14% (95.64%) the time on PC1 (PC2) when searching the four query sequences, respectively. For the pure GPU algorithm SwNw_GPU, the GTX 1070 graphics card on PC2 runs up to 4.36 times faster compared to the GTX 750 Ti graphics card on PC1.

Run time comparison results
Figure 8 shows the evaluation results in GCUPS from which we can observe that our parallel algorithms can reach higher GCUPS scores compared to the serial algorithm. When searching with the four query sequences, the heterogeneous parallel algorithm SwNw_MIX can reach the highest performance of 4.33 GCUPS and the lowest performance of 2.91 GCUPS on PC1, and the highest performance of 30.04 GCUPS and the lowest performance of 14.5 GCUPS on PC2, respectively.

Performance test results measured in terms of GCUPS
Figrue 9 shows the speedup results of the parallel algorithms SwNw_GPU and SwNw_MIX executed on PC1 and PC2, respectively. The run time of i3-3240 CPU on PC1 is used as the serial time to compute the speedup results. From the results we can see that SwNw_MIX can achieve top speedups of 19.99 (138.86) times run on PC1 (PC2) when searching with the WP_011236883 query sequence, it outperforms SwNw_GPU which its top speedups are 16.68 (71.36) times on PC1 (PC2). The algorithms can gain much better performance results tested on PC2 than PC1 mainly because the former equipped with more powerful CPU processor and graphics card.

Speedup results of the parallel algorithms
5 Conclusion and future work
In this paper, a GPU-based parallel algorithm for hybrid search of protein database is designed and implemented. The hybrid search approach is introduced by combining the Smith–Waterman local alignment and the Needleman–Wunsch global alignment algorithms into one process. This method utilize the GPU’s parallel computing power to search protein databases quickly; in addition, the heterogeneous parallelism is realized through coupling CPU multi-threaded operations into the GPU algorithm for maximizing the performance. The parallel algorithm is implemented with the CUDA parallel computing framework, and the Swiss-Prot database is used in our experiment. We use the ELL storage format and the cache on GPU cores to optimize our parallel algorithm to achieve higher compute and storage efficiency in the implement process. The results show that the parallel algorithm proposed in this paper can achieve a performance over 30 GCUPS and save exceeding 95% the run time compared to the serial algorithm, significantly improving the speed and convenience of protein database search.
In the future research, we will attempt to apply the parallel algorithm to searches of larger sequence databases and continue to make optimizations for achieving higher performance.
References
Mount DW (2004) Bioinformatics: sequence and genome analysis. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol. Biol 48(3):443–453
Smith TF, Waterman MS (1981) Identification of common molecular subsequences. J Mol Biol 147(1):195–197
Altschul SF, Madden TL, Schaffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Pearson WR (1990) Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol 183:63–98
Keckler SW, Dally WJ, Khailany B et al (2011) GPUs and the future of parallel computing. IEEE Micro 31(5):7–17
Nickolls J, Dally WJ (2010) The GPU computing era. IEEE Micro 30(2):56–69
Nvidia (2016) Geforce GTX 1070. Nvidia. http://www.geforce.com/hardware/10series/geforce-gtx-1070
Nvidia (2014) CUDA C programming guide v6.5. Nvidia. http://docs.nvidia.com/cuda/pdf/CUDA_C_Programming_Guide
AMD (2013) AMD accelerated parallel processing OpenCL programming guide. AMD. http://developer.amd.com/wordpress/media/2013/07/AMD_Accelerated_Parallel_Processing_OpenCL_Programming_Guide-rev-2.7
Nickolls J, Buck I, Garland M et al (2008) Scalable parallel programming with CUDA. Queue 6(2):40–53
Eddy SR (2004) Where did the BLOSUM62 alignment score matrix come from? Nat Biotechnol 22(8):1035–1036
Yong-xian Wang, Zheng-hua Wang (2011) Introduction to bioinformatics. Tsinghua University Press, Beijing
Gotoh O (1982) An improved algorithm for matching biological sequences. J Mol Biol 162(3):705–708
Polyanovsky VO, Roytberg MA, Tumanyan VG (2011) Comparative analysis of the quality of a global algorithm and a local algorithm for alignment of two sequences. Algorithms Mol Biol 6(1):1–12
Liu Y, Huang W, Johnson J et al (2006) Gpu accelerated smith-waterman. In: Alexandrov VN, van Albada GD, Sloot PMA, Dongarra J (eds) Computational ScienceCICCS 2006. Springer, Berlin, pp 188–195
Manavski SA, Valle G (2008) CUDA compatible GPU cards as efficient hardware accelerators for Smith-Waterman sequence alignment. BMC Bioinf 9(2):1
Khajeh-Saeed A, Poole S, Perot JB (2010) Acceleration of the Smith–Waterman algorithm using single and multiple graphics processors. J Comput Phys 229(11):4247–4258
Blazewicz J, Frohmberg W, Kierzynka M et al (2011) Protein alignment algorithms with an efficient backtracking routine on multiple GPUs. BMC Bioinf 12(1):1
Siriwardena TRP, Ranasinghe DN (2010) Accelerating global sequence alignment using CUDA compatible multi-core GPU. In: 2010 Fifth International Conference on Information and Automation for Sustainability, vol 2010, pp 201–206
Kirk DB, Wen-mei WH (2009) Programming massively parallel processors: a hands-on approach. Morgan Kaufmann, Burlington
Cook S (2012) CUDA programming: a developer’s guide to parallel computing with GPUs. Morgan Kaufmann, Waltham
Acknowledgements
The authors would like to thank all the reviewers for their precious comments. This paper is supported by the Shandong Provincial Natural Science Foundation, China (Grant No. ZR2015CL020).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhou, W., Cai, Z., Lian, B. et al. Protein database search of hybrid alignment algorithm based on GPU parallel acceleration. J Supercomput 73, 4517–4534 (2017). https://doi.org/10.1007/s11227-017-2030-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11227-017-2030-x