
Abstract
Point mutation of amino acids is a means used by biotechnologists to improve the performance of proteins. To study a point-mutated polypeptide, one requires its global minimum energy conformation. This conformation can be determined by molecular dynamics via Langevin's equations of motion. Molecular dynamics simulations belong to the most difficult problems to parallelize in a scalable manner. We provide a method for defining a special purpose 3D array processor architecture for the molecular dynamics simulation of point-mutated polypeptides. The architecture is derived from a spatial decomposition of a known conformation of the point-mutated polypeptide or the native conformation of the given protein. By using an approximation scheme for the deterministic forces, the interprocessor communication can be kept local. The architecture affords a simple distributed load balancer and is scalable. The computational workload of the array processor architecture to perform molecular dynamics simulations under realistic conditions is addressed. An example architecture is given by point-mutated penicillin amidase.
Similar content being viewed by others
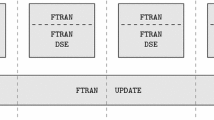

References
T.E. Creighton, Proteins: Structures and Molecular Principles, New York: Freemann, 1983.
M.P. Allen and D.J. Tildesley, Computer Simulation of Liquids, Oxford: Clarendon Press, 1987.
A.R. Leach, Molecular Modelling: Principles and Application, Addison Wesley, 1996.
B. Brooks, R. Bruccoleri, B. Olafson, D. States, S.Swaminathan, and M. Karplus, "CHARMM: A Program for Macromolecular Energy Minimization and Dynamics Calculations," Dept. of Chemistry, Harvard Univ., 1982.
A. Lyubartsev and A. Laaksonen, "MDYNAMIX-A Scalable Portable Parallel MD Simulation Package for Arbitrary Molecular Mixtures," Comp. Phys. Comm., vol. 128, 2000, pp. 565-589.
S.J. Weiner, P.A. Kollman, D.A. Case, U.C. Singh, C. Ghio, G. Alagona, S. Profeta, and P. Weiner, "A New Force Field for Molecular Mechanical Simulation of Nuclear Acids and Proteins," J. Am. Chem. Soc., vol. 106, 1984, pp. 765-784.
R.K. Brunner, J.C. Phillips, and L.V. Kale, "Scalable Molecular Dynamics for Large Biomolecular Systems," Preprint Dept. Computer Science, Urbana Univ, Il, 2000.
M. Nelson, W. Humphrey, A. Gursoy, A. Dalke, L. Kale, R.D. Skeel, and K. Schulten, "NAMD-A Parallel Object-Oriented Molecular Dynamics Program," Int. J. Supercomput. Applic. High Performance Computing, vol. 10, 1996, pp. 251-268.
S. Plimpton and B. Hendrickson, "A New Parallel Method for Molecular Dynamics Simulation of Macromolecular Systems," J. Comput. Chem., vol. 17, 1996, pp. 326-337.
M. Eichinger, H. Grubm¨uller, and H. Haller, "User Manual for EGO VIII (Release 2.0)," Leibniz Rechenzentrum, Munich, 2000.
C.B. Anfinsen, E. Haber, M. Sela, and F.H. White Jr., Proc. Natl. Acad. Sci., vol. 47, 1961, pp. 1309-1314.
G. Nemethy, M.S. Pottle, and H.S. Scheraga, "Energy Parameters in Polypeptides. Updating the Geometric Parameters, Nonbonded Interactions and Hydrogen Bond Interactions for the Naturally Occurring Amino Acids," J. Phys. Chem., vol. 87, 1983, pp. 1883-1887.
A. Liwo, "AUnited-Residue Force Field for Off-Lattice Protein-Structure Simulations. I. Functional Forms and Parameters of Long-Range Side-Chain Interaction Potentials from Protein Crystal Data," J. Comp. Chem., vol. 18, p.6.
J.L. Klepeis and C.A. Floudas, "Deterministic Global Optimization and Torsion Angle Dynamics for Molecular Structure Prediction," Techn. Report, Dept. Chem. Eng., Princeton Univ., Princeton, NJ.
A. Neumaier, "Molecular Modelling of Proteins and Mathematical Prediction of Protein Structure," SIAM Rev., vol. 39, 1997, pp. 407-460.
H.A. Scheraga, "Recent Developments in the Theory of Protein Folding: Searching for the Global Energy Minimum," Biophys. Chem., vol. 59, 1996, pp. 329-339.
W.F. van Gusteren and H.J.C. Berendsen, "Computer Simulation of Molecular Dynamics: Methodology, Applications, and Perspectives in Chemistry," Angew. Chemie Int. Ed. in English, vol. 29, 1990, pp. 992-1023.
L. Hewitt, V. Kasche, K. Lummer, R.J. Lewist, G.N. Marshudov, C.S.Verma, G.G. Dodson, and K.S.Wilson, "Structure of a Slow Processing Precursor Penicillin Acylase from Escherichia coli Reveals the Linker Peptide Blocking the Active Site Cleft," J. Mol. Biology, vol. 302, 2000, pp. 887-898.
V. Kasche, K. Lummer, A. Nurk, E. Piotraschke, A. Rieks, S. Stoeva, and W. Voelter, "Intramolecular Proteolysis Initiates the Maturation of Penicillin Amidase from E. coli. Biochem. Biophys. Acta, vol. 1433, 1999, pp. 76-86.
L. Hewitt, V. Kasche, K. Lummer, R.J. Lewist, G.N. Marshudov, C.S.Verma, G.G. Dodson, and K.S.Wilson, "ASlowProcessing Precursor Penicillin Acylase from Escherichia coli," PDB ID: 1E3A, PDB, 2000.
K. Lummer, "Inter-und intramolekulare enzymatische Katalysierte Reaktionen am Beispiel der Penicillinamidase,"Dissertation, TU Hamburg-Harburg, 2000.
K.-H. Zimmermann, T. Lai, Z. Ignatova, B. Galunsky, and V. Kasche, "Prediction of Point-Mutated Penicillin Amidase Precursor from Escherichia coli via Simulated Annealing," Preprint, 2002.
K. Lehmann, "Molecular Dynamics of Proteins," Diploma Thesis, TU Hamburg-Harburg, 2002.
S. Kirkpatrick, C.D. Gelatt, and M.P. Vecchi, "Optimization by Simulated Annealing," Science, vol. 220, 1983, pp. 671-680.
S.Y. Kung, VLSI Array Processors, Prentice Hall, 1987.
K.-H. Zimmermann, "Linear Mappings of nDRecurrence Equations onto kD Array Processors," J. VLSI Signal Processing, vol. 12, 1996, pp. 187-202.
K. Jainandunsing, "Optimal Partitioning Scheme for Wavefront/ Systolic Array Processors," Proc. IEEE Symp. on Circuits and Systems, 1986, pp. 940-943.
K.-H. Zimmermann, "A Unifying Lattice-Based Approach for the Partitioning of Systolic Arrays via LPGS and LSGP," J. VLSI Signal Processing, vol. 17, 1997, pp. 21-42.
M. Markiewicz, "Dipole Moments of Protein Structures," Project Work, TU Hamburg-Harburg, 2002.
N.A. Lynch, Distributed Algorithms, San Francisco: Morgan Kaufmann, 1996.
H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H.Weissig, I.N. Shindyalov, and P.E. Bourne, "The Protein Data Bank," Nucleic Acids Res., vol. 28, 2000, pp. 235-242.
Intel Itanium 2: www.intel.com
H. El-Rewini and T.G. Lewis, Distributed and Parallel Computing, Manning, Greenwich, CT, 1997.
T.T. Lai, "Implementing a Simulated Annealing Algorithm for the Prediction of Point-Mutated Penicillin G Acylase," Master Thesis, TU Hamburg-Harburg, 2001. A Special Purpose Array Processor Architecture 309
N. Metropolis, A.W. Rosenbluth, A.H. Teller, and E. Teller, "Equation of State Calculation by Fast Computing Machines," J. Chem. Physics, vol.21, 1953, pp. 1087-1091.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Zimmermann, KH. A Special Purpose Array Processor Architecture for the Molecular Dynamics Simulation of Point-Mutated Proteins. The Journal of VLSI Signal Processing-Systems for Signal, Image, and Video Technology 35, 297–309 (2003). https://doi.org/10.1023/B:VLSI.0000003027.47559.1e
Published:
Issue Date:
DOI: https://doi.org/10.1023/B:VLSI.0000003027.47559.1e