





Abstract
Transcription and DNA supercoiling are involved in a complex, dynamical and non-linear coupling that results from the basal interaction between DNA and RNA polymerase. We present the first software to simulate this coupling, applicable to a wide range of bacterial organisms. TwisTranscripT allows quantifying its contribution in global transcriptional regulation, and provides a mechanistic basis for the widely observed, evolutionarily conserved and currently unexplained co-regulation of adjacent operons that might play an important role in genome evolution.
TwisTranscripT is freely available at https://github.com/sammeyer2017/TwisTranscripT. It is implemented in Python3 and supported on MacOS X, Linux and Windows.
1 Introduction
Transcription is involved in a complex dynamical coupling with DNA supercoiling (SC), i.e. the mechanical distortions of the double-helix induced by torsional stress. On the one hand, negative SC strongly facilitates the opening of the double-helix during transcription initiation, and thereby acts as a basal transcriptional regulator (Travers and Muskhelishvili, 2005). Conversely, the rapid rotation of the RNA polymerase (RNAP) imposed by the double-helical geometry during transcription elongation is hindered by the viscosity and crowding of the medium, resulting in an asymmetrical accumulation of torsional stress, from back to front. The resulting mechanical deformation quickly propagates at kilobase distances and gives rise to a complex, orientation–dependent interaction between adjacent genes (El Houdaigui et al., 2019). This coupling affects eukaryotes as well as prokaryotes (Gilbert and Allan, 2014; Travers and Muskhelishvili, 2005), albeit with different rules owing to differences in topoisomerase enzymes and genome architectures. In the following, we focus on bacteria, where transcriptional regulation by SC was studied more extensively.
Several theoretical models of the coupling were developed recently. SC has been described to regulate transcription at the initiation step (Brackley et al., 2016), accounting for dynamical features of transcription observed in vitro (Chong et al., 2014). However, that model does not incorporate in vivo relevant components such as topoisomerases, which are required to reproduce many experimental assays, e.g. inhibition of the latter by antibiotics. Another model is based on the stalling action of positive SC on transcription elongation (Sevier and Levine, 2018), which in vivo is mostly relevant to highly transcribed genes such as those of ribosomal RNAs. The model proposed here combines a regulatory action of SC during transcription initiation relevant to most moderately expressed bacterial genes, with an explicit description of topoisomerases. As a result, it is possible to simulate many in vivo experimental assays, in particular transcriptomes obtained after topoisomerase inhibition or environmental stress (El Houdaigui et al., 2019). This constitutes an important step toward quantifying the contribution of SC in the global regulation of bacterial transcription. The reader should, however, keep in mind that SC affects transcription at several stages of the process and through complex mechanisms (Martis et al., 2019), most of which are treated in a strongly simplified way; as a result, simulations of transcription from specific or unusual promoters might exhibit discrepancies with experimental results.
We propose an implementation of this model in Python, which constitutes the first available simulation software of the transcription-SC coupling. In contrast to classical regulation based on promoter-specific transcription factors, the latter regulation mode results from the basal activity of RNAP itself and is, therefore, relevant to most genomic loci in a wide range of bacterial species. It constitutes an attractive explanation for the coregulation of adjacent genes widely observed in bacteria and currently unexplained (Junier and Rivoire, 2016), in particular, within so-called pathogenicity islands (Martis et al., 2019).
2 Implementation
The code is available at https://github.com/sammeyer2017/TwisTranscripT. It was written in Python3 and relies on the NumPy (Van Der Walt et al., 2011) and Pandas libraries. It simulates stochastic transcription along a 1D genome for a prescribed time duration with the Euler algorithm. An input configuration file (in standard format) must be provided, with a list of genomic annotation files and simulation parameters, as follows.
Four annotation files describe the simulated genome, which can be an entire chromosome/plasmid or a limited chromosomal region: standard gene annotation file (GFF); list of transcription start site (TSS) and termination sites (TTS) positions, directions and strengths (initiation of stochastic rates and termination probabilities, respectively); list of fixed topological barrier positions (BARR_FIX). Imperfect termination gives rise to partial co-expression of successive isodirectional genes by stochastic transcriptional read-through, which is widespread in bacteria (Conway et al., 2014). Note that overlapping transcription units (alternate TSSs or overlapping antisense transcripts) are not accepted in the present version because of possible RNAP clashes.
Simulation parameters are separated into five categories. In most cases, only the first and last ones should be modified, whereas other parameters are generic and calibrated for bacterial transcription, with equations and methods described in detail in El Houdaigui et al. (2019):
INPUTS: paths of the four files above.
PROMOTER: parameters of the promoter response curve to local SC variations, , where the opening free energy U follows a sigmoidal curve based on DNA thermodynamics: . Default values [, m = 2.5, see Figure 1C in El Houdaigui et al. (2019)] are valid for many bacterial genes investigated in vitro and must be adapted for unusual promoters activated by DNA relaxation such as gyrA. The dependence might, however, be too simplistic for specific promoters subject to alternate effects such as complex DNA structural transitions.
TOPOISOMERASES: parameters of SC-dependent topoisomerase activity, following sigmoidal curves (equation above) with default values calibrated for bacteria (topoisomerase 1 and DNA gyrase): see Figure 1D in El Houdaigui et al. (2019).
GLOBAL: space and time discretization units. Default values: 60 nt and 2 s.
SIMULATION: Topoisomerase effective concentrations in μM, SC generation rate by elongating RNAPs (default 0.2 coils per unit length), initial uniform SC level, number of RNAP copies, total simulation time (in seconds) and time interval between exports. Note that an ‘effective concentration’ is used to quantify the ATP-dependent gyrase activity; it is proportional to the concentration of the ATP-gyrase complex and was calibrated on an in vitro assay (Chong et al., 2014). Tuning this effective concentration allows mimicking the in vivo action of gyrase inhibitors or environmental stresses affecting the ATP/ADP ratio.
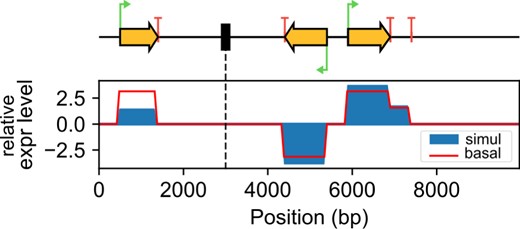
Illustration of the activating effect of transcription-generated negative supercoiling in a pair of divergent promoters (right), whereas another promoter (left) is inhibited by positive torsion generated downstream (the genome is circular). Red line: transcript coverage based on basal initiation rates; blue areas: coverage affected by TSC from a simulation. The central black rectangle represents a topological barrier. The third gene has an imperfect terminator (of strength 0.5), followed by a 3′-untranslated region before reaching a perfect terminator. (Color version of this figure is available at Bioinformatics online.)
Output files are of three types (see details in the program description):
csv files with the list of possible transcripts, and their expression levels and times during the simulation.
all_res directory with detailed information exported at regular time intervals during the simulation: position of RNAPs, transcripts and SC distributions.
resume_sim directory with files required to restart a previous simulations (possibly with different parameters).
3 Conditions of use
Figure 1 illustrates a simulation involving a small genomic region. The regulatory effect of the coupling can be analyzed by comparing the basal expression profile based on TSS/TTS strengths with the observed profile where initiation rates are dynamically modulated by transcription-coupled supercoils (both profiles are normalized). Because of stochasticity, a quantitative analysis of the mechanism requires multiple simulations and a subsequent statistical analysis of the expression profiles (El Houdaigui et al., 2019). An additional plotting package is provided with some useful functions (e.g. an automatic representation of the genome as in Fig. 1), which rely on SciPy, MatPlotLib, DNAplotlib libraries, but each analysis usually requires a specific statistics/plotting code.
The program was mostly applied on genomic regions of <100 kb, in which case a typical simulation takes <1 min on a standard laptop computer. It can be applied on a whole chromosome, but it is not recommended since topological domains separated by fixed barriers are independent and can thus usually be simulated separately with a lower computational load. The computation time increases linearly with the simulated time, and also depends essentially on the number of topological boundaries (transcribing RNAPs or fixed barriers).
The software was shown to reproduce the regulatory action of SC observed in specific in vitro or in vivo transcription assays involving model promoters, as well as orientation-dependent response of most bacterial genes to SC variations (El Houdaigui et al., 2019). It may be inaccurate for highly transcribed operons such as those of ribosomal genes, where positive SC accumulates and gyrase is recruited in a sequence-specific manner. Conversely, it seems particularly relevant for analyzing the co-expression of adjacent genes lacking any common regulator, as observed in transcriptomics data in many genomic loci (Junier and Rivoire, 2016), in particular within pathogenicity islands (Martis et al., 2019).
Such simulations require appropriate input files, which raises the following main difficulties: (i) transcription units are not always well-defined in bacteria, due to ‘pervasive transcription’ (Conway et al., 2014) that may contribute to the coupling; (ii) transcription initiation rates can only be semi-quantitatively inferred from transcriptomics data, due to normalization steps, inhomogeneous transcript degradation rates and coverage; (iii) topological barriers are currently not well-defined in bacterial chromosomes, and their definition in simulations partly relies on arbitrary choices, the effect of which must be carefully analyzed; (iv) finite-size effects must be equally checked for (number of RNAP molecules, simulation time).
Although the simulation code was developed and calibrated in bacteria, it might be adapted to eukaryotes where a similar coupling occurs (Gilbert and Allan, 2014). This extension would involve (i) calibrating an effective promoter response curve to variations in local SC (which could reach positive as well as negative values), as observed in yeast (Meyer and Beslon, 2014) and (ii) adapting the equations of topoisomerase activities to eukaryotic type-1 and type-2 enzymes. Since transcriptional regulation by SC might involve different mechanisms in eukaryotes (Valdes et al., 2019) (because of positive SC levels, nucleosomes, etc.), the application range and predictive power of the model should then be tested.
Funding
This work was supported by grants from INSA Lyon [BQR 2016]; and Agence Nationale de la Recherche [ANR-18-CE45-0006-01 to S.M.].
Conflict of Interest: none declared.
References


{kind=link}