
Abstract
Institutions that specialize in prostate MRI acquire different MR sequences owing to variability in scanning procedure and scanner hardware. We propose a novel prostate cancer detector that can operate in the absence of MR imaging sequences. Our novel prostate cancer detector first trains a forest of random ferns on all MR sequences and then decomposes these random ferns into a sum of MR sequence-specific random ferns enabling predictions to be made in the absence of one or more of these MR sequences. To accomplish this, we first show that a sum of random ferns can be exactly represented by another random fern and then we propose a method to approximately decompose an arbitrary random fern into a sum of random ferns. We show that our decomposed detector can maintain good performance when some MR sequences are omitted.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


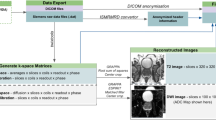
1 Introduction
The use of multi-parametric MRI (mpMRI) is the most effective way to detect and biopsy prostate cancer [4]. However, institutions specializing in prostate MRI have different scanning procedures and hardware resulting in different MR images. This particularly poses a challenge for computer-aided detection (CAD) of prostate cancer as many existing CAD methods (e.g. [7, 9, 12]) were developed for specific MR sequences coming from a single institution and will not function in the absence of expected MR images. To address this problem, we propose a novel prostate CAD that is capable of making predictions in the absence of one or more MR sequences it was trained on. This might include sequences like Dynamic Contrast Enhancement (DCE) or high b-value (e.g. B1500) diffusion images that may not be acquired owing to patient comfort or limitations of scanner hardware respectively. Our CAD uses T2 weighted (T2W), apparent diffusion coefficient (ADC) and B1500 MR sequences and first builds several random ferns that use features computed on T2W, ADC and B1500 images combined. Then we present a method to decompose the trained random ferns into a sum of random ferns that each individually operate on one of T2W, ADC and B1500 images. The result is a prostate CAD that can simply exclude MR sequence-specific models from the sum during prediction. We show that the decomposed model exhibits similar performance to the original model. We also explore the performance consequences of missing information. Furthermore, we show that our prostate CAD can maintain good performance when omitting several combinations of MR sequences.

Examples of T2W, ADC and calculated B1500 images with corresponding CAD probability map. Hypointense regions in T2W and ADC can indicate the presence of prostate cancer, while hyperintense regions in B1500 indicate the presence of prostate cancer. Probability map colors range from blue (low score) to red (high score). There is a clinically significant (Gleason 3 + 4 or higher) transition zone lesion in this example that is correctly predicted by the CAD.
2 Related Works
There are several examples of prostate CAD systems in the literature. The general workflow for these systems is to read in several images from mpMRI and then produce a colorized confidence or probability map indicating voxel-by-voxel suspicious regions. Some systems [9] further process these probability maps to extract lesion segmentations. The probability maps can then be interpreted by radiologists or used in guided biopsy procedures. Figure 1 shows a few examples of mpMRI images and probability maps. A few recent examples of CAD include the works of [7, 9, 12]. The system in [7] uses Support Vector Machine (SVM) with local binary pattern features computed on T2W and B2000 images. The work of [9] uses random forest, Gentle Boost, and LDA each using a combination of T2W, ADC, B800 and DCE images with features based on pixel intensity, relative location in the prostate, texture, blobness and pharmacokinetic heuristics. This work further segments and classifies lesions based on the output from the voxel classifier. The system proposed in [12] picks candidate locations based on a multi-scale Hessian-based blobness filter which are then each classified with an LDA classifier. The LDA classifier uses statistics-based features computed on T1W, T2W, ADC, DCE and Ktrans images. Although not applied to mpMRI or prostate cancer detection, the work of [6] develops a deep learning system that can train and test in the absence of images. Their system uses a set of disjoint neural network (NN) pipelines that individually process different kinds of images. The output of these pipelines are then averaged to produce the prediction. When an image is missing, the average omits the output from the corresponding pipeline.
Our system is fundamentally different to existing prostate CAD systems in that it does not require the availability of all MR sequences used to train the model and can still make predictions even if operating on a single image. Other differences to existing prostate CAD systems include the use of a different classifier, the use of a transition zone segmentation, as well as the use of different features. We additionally introduce a new way to train random ferns and a way to decompose them into a sum of random ferns. While the work of [6] can also operate in the absence of images, our method infers sequence-specific classifiers through an explicit model decomposition while [6] jointly optimizes the modality-specific pipelines in an end-to-end fashion.
3 Methods
The proposed prostate CAD operates on 2D image slices and is comprised of pixel-wise random fern classifiers [11] employing intensity statistics and Haralick texture features [5]. The random ferns are first trained on features calculated on all T2W, ADC and B1500 MR images and then the resulting model is decomposed into a sum of random ferns that each operate individually on features calculated on T2W, ADC and B1500. Figure 1 shows an example of these MR sequences and corresponding CAD prediction. When one or more of these MR sequences are missing, the CAD excludes the corresponding random ferns from the evaluation and can still produce a probability map. We chose random ferns since they are similar to and simpler than decision trees and are thus robust and powerful learners while being intuitive to understand and manipulate.
3.1 Random Ferns
Random ferns [11] are constrained decision trees that behave like checklists of yes/no questions based on the input feature vector. The combination of yes/no answers are then used to lookup a prediction. As observed in [11], this checklist behavior is synonymous to a decision tree using the same decision criteria at each level of the tree.
Our method modifies random ferns in a number of ways. First, the binary decisions for each fern are selected by employing Feature Selection and Annealing (FSA) [2]. Second, we use mean aggregation instead of semi-naïve Bayes aggregation. Several independent random ferns are trained on randomly sampled data to produce a forest of random ferns.
Where decision tree training employs a simple recursive optimization strategy to select binary decisions, the constrained decision structure of random ferns make such recursive optimization strategies prohibitively expensive. We avoid this issue entirely by instead noting that a sum of decision stumps can be exactly represented as a random fern as illustrated in Fig. 2. We instead optimize a sum of decision stumps and assume the resulting model is approximately the unknown optimal random fern.
To select optimal binary decisions for the random fern, decision stumps are first exhaustively generated for each feature. This is done by discretizing the range of observed feature responses and using these as decision thresholds for each decision stump. Then a coefficient is associated with each leaf of every stump. The sum of these stumps forms a linear model for FSA to optimize. FSA simultaneously minimizes a loss function while selecting the most informative decision stumps. As in [2], we use the Lorenz loss function for FSA in this work. Lastly, the FSA-selected decisions are used to train the fern by passing examples down the fern and computing the label distribution \(p(y|\mathbf {x})\) in each leaf where \(y \in \{ 0, 1 \}\) is the class label and \(\mathbf {x}\in \mathbf {R}^F\) is the feature vector. This process is repeated on several randomly sampled training sets to form a forest of jointly trained random ferns.

Example of a sum of two decision stumps resulting in a random fern. This generalizes when summing more than two stumps. Note that an arbitrary random fern cannot be exactly represented by a sum of stumps in general.
3.2 Random Fern Decomposition
Each jointly trained random fern T is then decomposed into three MR sequence-specific random ferns by first forming empty random ferns each using only T2W, ADC and B1500 binary decisions from the original random fern. More specifically, if L represents a leaf of the original depth D random fern T with \(L \in T\), then L represents a unique combination of yes/no answers that identify that specific leaf. We then augment \(L = \{ \ell _1\ |\ \ell _2\ |\ \ell _3 \}\) where \(\ell _m\) represents both a leaf of a random fern \(T_m\) specific to MR sequence m (T2W: \(m=1\), ADC: \(m=2\), B1500: \(m=3\)) as well as the outcome of decision criteria specific to that MR sequence. We additionally use the notation \(I(\ell \subseteq L) \in \{0, 1\}\) to indicate that a subset of the decision criteria outcomes \(\ell \) match in L. We also further abuse notation by treating leaves \(L \in T, \ \ell \in T_m\) as integers. Then we use specialized Artificial Prediction Market (APM) [1] to fit the sum of three MR sequence-specific random ferns to the jointly trained random fern. We chose specialized APM since it supports models that can abstain from participation (e.g. due to missing information). We treat random fern T as the ground truth and can thus exactly calculate the mass p(L) of examples occurring in leaf L as well as the label distribution p(y|L) in leaf L. We can then indirectly infer a linear aggregation c(y|L) of the sequence specific random ferns \(T_1,T_2,T_3\) by iterating the update rule (2) over each populated leaf \(L \in T\).
Here Z is a normalizer, \(\eta \) is the learning rate, \(\beta _{m, \ell , y}\) the learned prediction of leaf \(\ell \). The update rule is repeatedly used for each leaf L from the jointly trained random fern until convergence. The result is 3 constituent random ferns with leaves \(\ell \) predicting \(\beta _{m, \ell , y}\). The resulting ferns can then be aggregated using a form similar to c(y|L) as the predicted probability and this is given as
where Z is a normalizer, \(\ell _m = T_m(\mathbf {x})\) is the predicted leaf for feature vector \(\mathbf {x}\) on MR image \(I_m\).
3.3 Data
We train and evaluate the proposed method on a combination of cases from NIH and data made available through the recent ProstateX Challenge [9]. All MR sequences (T2W, ADC, B1500) were aligned, resampled to \(0.35\,\text {mm} \times 0.35\,\text {mm} \times 3\,\text {mm}\), and normalized using a Rician-based normalization [8]. Cases from NIH feature 19 healthy patients and 49 pathology-corroborated hand-drawn lesions prepared by a radiologist. The ProstateX data featured a database of points with corresponding clinical significance binary label for 204 training cases.
3.4 Prostate Segmentation
This CAD relies on the presence of a prostate and transition zone segmentation as it trains separate transition zone and peripheral zone classifiers. Segmentations for the NIH cases were prepared manually by a radiologist while the 204 ProstateX cases were automatically segmented by an algorithm based on [3, 10] with the possibility of manual correction. The method does depend on a well bounded prostate segmentation since it is liable to misclassify the background in T2W or ADC.
3.5 Training
Each fern was trained on an independent random subset of training cases without regard to annotation type. Positive and negative points were densely sampled inside the prostate for healthy cases and cases with hand-drawn lesion contours. Positives were densely sampled within 5 mm of clinically significant points in ProstateX cases. No negatives were sampled from ProstateX cases since lesion extent is not known and the reason for the points in the database are not known.
3.6 Statistical Analysis
The proposed CAD was analyzed on five sets of two fold cross validation experiments with each cross validation experiment generated on randomly shuffled data and thus resulting in 10 total distinct experiments. Our 10 experiments always train and test on \({\approx }1/2\) and \({\approx }1/2\) of data set and we believe this better characterizes the generalizability of the method than either 2 fold or 10 fold cross validation. Cross validation was used purely for assessing performance and no hyperparameters were picked using the test folds. The data were split so that approximately the same number of healthy, contour and ProstateX cases were used in each fold. We compared the original random fern model to the decomposed random fern model. We also considered the performance of the proposed CAD on a variety of combinations of T2W, ADC, and B1500 images. Performance was measured in terms of ROC curves for detection and clinical significance classification.
To demonstrate the difference between decomposing MR sequence-specific models and training directly on MR sequences we retrained the decomposed T2W, ADC and B1500 models on examples coming from each MR sequence. For the purpose of comparing the models, the decision criteria were kept constant and the fern predictions were directly recalculated on its training set. The resulting AUCs and margins were calculated for the two models over the five permutations of two fold cross validation. The margin was calculated in a weighted sense and is given by
where \(X^+\) and \(X^-\) are the positive and negative examples and \(| \cdot |\) denotes cardinality.
Detection ROC. Detections and false positives were determined on the cases with lesion contour annotations. Probability maps and contour annotations were first stacked to define 3D probability maps and lesion masks. For each lesion, the 90th percentile of the probability scores occurring inside the lesion was taken to be the lesion’s score. If the lesion score exceeded a threshold, then the lesion was said to be detected. False positives were determined by cutting the prostate into \(3\,\text{ mm } \times 3\,\text{ mm } \times 3\,\text{ mm }\) cubes. The 90th percentile of probabilities occurring inside each cube were determined and used as the cube’s score. If a cube did not coincide with a lesion and its score exceeded a threshold, then cube was said to be a false positive.
ProstateX ROC. The ProstateX Challenge data includes an annotated database of image points and whether they correspond to clinically significant cancer or not. Each point was scored by the CAD by first stacking the probability maps into a 3D probability map. Then the 90th percentile of probability scores occurring inside a 5 mm ball was taken to be the point’s score. A classification ROC curve was then calculated against these scores and their ground truth.
4 Results
The ROC curves were calculated on each of the test sets from each of the cross validation experiments and were averaged with respect to false positive rate. These averaged ROC curves are displayed in Fig. 3. The curves compare the performance of the CAD using the random ferns trained on all MR sequences (Jointly Trained) to the CAD using decomposed random ferns evaluated on a combination of MR sequences.
Table 1 features AUCs and margins of the T2W, ADC and B1500 decomposed models and their retrained counterparts while Fig. 4 illustrates the margin’s effects on the decomposed T2W model and retrained T2W model.

ROC curves of the CAD system evaluated as a detector and a clinical significance classifier.

An example of probability maps produced by the decomposed model using T2W+ADC+B1500 (a), the T2W model (b) and the retrained T2W model (c). The latter illustrates the reported low margin of the retrained model.
5 Discussion
The detection ROC in Fig. 3 reveals similar performance between the jointly trained model and the decomposed model using all sequences (T2W+ADC+B1500) demonstrating no loss in detection performance even after decomposing the jointly trained model. When some sequences are missing, the CAD is able to maintain similar or good detection performance with decomposed models ADC+B1500, T2W+B1500, ADC and B1500 achieving not less than 0.9 AUC in performance. When diffusion is completely excluded, we see that the decomposed CAD can still achieve a good AUC of 0.84, the lowest of all reported detection AUCs.
Similar ProstateX performance is also seen in Fig. 3 between the jointly trained model and decomposed model again showing little to no loss in performance. The ProstateX data set is comprised of outcomes of targeted biopsies which implies that the data set may be biased toward only suspicious prostate regions and many false positives such as artifacts or benign structures are likely to be already ruled out. For this reason and based on findings in the work of [7], it is not beyond expectation to find that B1500 achieved the highest ProstateX AUC of 0.84.
Table 1 shows the value of decomposition over training single sequence models. Importantly, both single-sequence models use identical features and decision criteria for fair comparison. While the detection and ProstateX AUCs of the two models are similar, the prediction margin of the decomposed models are higher and would produce more contrasting probability maps as seen in Fig. 4.
Lastly, owing to differing evaluation methodology, private data sets, and the lack of prostate CAD systems that can operate in the absence of MR sequences, it is difficult, perhaps even meaningless, to objectively compare our performance with other methods in the literature. However, this method did place third in the ProstateX competition with a test AUC of 0.83 tailing methods that achieved test AUCs of 0.84 and 0.87.
6 Conclusion
Decomposing random ferns to operate on individual MR sequences provides increased flexibility with little to no performance loss when working with data that may or may not include some MR sequences. Many combinations of MR sequences were also shown to provide similar performance to the CAD using all available MR sequences. The decomposed models also generally provide more contrasting positive and negative predictions while matching the performance of same models explicitly retrained on individual sequences.
References
Barbu, A., Lay, N.: An introduction to artificial prediction markets for classification. JMLR 13(Jul), 2177–2204 (2012)
Barbu, A., She, Y., Ding, L., et al.: Feature selection with annealing for computer vision and big data learning. IEEE TPAMI 39(2), 272–286 (2017)
Cheng, R., Roth, H.R., Lay, N.S., et al.: Automatic magnetic resonance prostate segmentation by deep learning with holistically nested networks. JMI 4(4), 041302 (2017)
Costa, D.N., Pedrosa, I., Donato Jr., F., et al.: MR imaging-transrectal us fusion for targeted prostate biopsies: implications for diagnosis and clinical management. Radiographics 35(3), 696–708 (2015)
Haralick, R.M., Shanmugam, K., et al.: Textural features for image classification. IEEE T-SMCA 3(6), 610–621 (1973)
Havaei, M., Guizard, N., Chapados, N., Bengio, Y.: HeMIS: Hetero-Modal Image Segmentation. In: Ourselin, S., Joskowicz, L., Sabuncu, M.R., Unal, G., Wells, W. (eds.) MICCAI 2016. LNCS, vol. 9901, pp. 469–477. Springer, Cham (2016). https://doi.org/10.1007/978-3-319-46723-8_54
Kwak, J.T., Xu, S., Wood, B.J., et al.: Automated prostate cancer detection using T2-weighted and high-b-value diffusion-weighted magnetic resonance imaging. Med. Phys. 42(5), 2368–2378 (2015)
Lemaître, G., Rastgoo, M., Massich, J., et al.: Normalization of T2W-MRI prostate images using Rician a priori. In: Medical Imaging 2016: Computer-Aided Diagnosis, vol. 9785, p. 978529. SPIE (2016)
Litjens, G., Debats, O., Barentsz, J., et al.: Computer-aided detection of prostate cancer in MRI. IEEE TMI 33(5), 1083–1092 (2014)
Nogues, I., et al.: Automatic Lymph Node Cluster Segmentation Using Holistically-Nested Neural Networks and Structured Optimization in CT Images. In: Ourselin, S., Joskowicz, L., Sabuncu, M.R., Unal, G., Wells, W. (eds.) MICCAI 2016. LNCS, vol. 9901, pp. 388–397. Springer, Cham (2016). https://doi.org/10.1007/978-3-319-46723-8_45
Ozuysal, M., Calonder, M., Lepetit, V., et al.: Fast keypoint recognition using random ferns. IEEE TPAMI 32(3), 448–461 (2010)
Vos, P., Barentsz, J., Karssemeijer, N., et al.: Automatic computer-aided detection of prostate cancer based on multiparametric magnetic resonance image analysis. PMB 57(6), 1527 (2012)
Acknowledgements
This research was funded by the Intramural Research Program of the National Institutes of Health, Clinical Center. Data used in this research were obtained from The Cancer Imaging Archive (TCIA) sponsored by the SPIE, NCI/NIH, AAPM, and Radboud University.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this paper

Cite this paper
Lay, N. et al. (2018). A Decomposable Model for the Detection of Prostate Cancer in Multi-parametric MRI. In: Frangi, A., Schnabel, J., Davatzikos, C., Alberola-López, C., Fichtinger, G. (eds) Medical Image Computing and Computer Assisted Intervention – MICCAI 2018. MICCAI 2018. Lecture Notes in Computer Science(), vol 11071. Springer, Cham. https://doi.org/10.1007/978-3-030-00934-2_103
Download citation
DOI: https://doi.org/10.1007/978-3-030-00934-2_103
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-00933-5
Online ISBN: 978-3-030-00934-2
eBook Packages: Computer ScienceComputer Science (R0)