
Abstract
The development of the next generation sequencing technology (NGS) has advanced the genomics research in many application domains. Metagenomics is one such powerful approach to study large community of microbial species. For the unknown species in the metagenomic samples, gene assembly and identification without a reference genome is a very challenging problem. To overcome this issue, distributed gene assembly software handling multiple metagenome samples can be used. In this paper, based on our previously developed highly scalable gene assembly software SWAP, we present a work flow called WFswap to assemble large genomic data based on many samples and to identify more genes. Our results suggested that WFswap is able to identify 94.2% of the bench-mark genes when tested on the 19 metagenomic samples that contain Bifidobacterium animalis subsp. lactis CNCM I-2494. Our proposed work-flow WFswap showed better performance than WFsoap, a similar workflow that used SOAPdenovo2 for gene assembly.
Z. Zheng and N. Guo—Equal Contribution.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Background
The outcome of Next Generation Sequencing technologies (NGS) is featured by very short fragments and extremely high throughput. NGS technology can provide valuable biological information by sequencing DNA fragments of any species. With these outstanding features such as high throughput, NGS provides a broad platform for researchers in biological sciences. For the past few decades, NGS has been widely used in many research domains and application areas, such as metagenomics. For the species that lack reference genomes, de novo genome assembly is the first and most fundamental step for downstream analysis [1]. For example, researchers use de novo assembly for discovering variation by aligning sequencing data to a reference sequence.
The reference sequences can be assembled with the help of various assembly methods. The methods such as SSAKE [2], VCAKE [3] and SHARGCS [4] use greedy algorithm to find pairs of reads with a large amount of overlaps and then merge them into longer sequences. These methods based on greedy algorithm are not efficient on genomes with more repeat regions [5]. Arachne [6], Celera Assembler [7], CAP3 [8], PCAP [9], Phrap [10], Phusion [11] and Newbler [12] adopt Overlap-Layout-Consensus (OLC) as the assembly algorithm. Overlap-Layout-Consensus algorithm builds the simplified overlap graph for generating contigs and is more suitable for the Sanger sequencing technology. While for NGS technology, de Bruijn graph [13] based assembly methods are widely used to assemble very short reads (much shorter than the ones from the Sanger sequencing technology).
SOAPdenovo2 [14] is a popular assembly software based on de Bruijn graph technology and can only run on a single computing node, hence the memory of the computing node limits its ability for assembling large dataset. IDBA-UD [15] is designed for sequences of single cell and metagenome. Megahit [16] can generate a large number of contigs, and is suitable for the assembly of complex samples (such as soil and water samples). For genomic data from massive parallel sequencing machines, the ability of genome assemblers to analyze these huge datasets plays a key role in genomic research. In our previous work, we developed a powerful scalable genome assembler called SWAP-Assembler 2 which runs on thousands of cores [17]. SWAP-Assembler 2 can assemble the Yanhuang genome dataset [18] in 26 min using 2,048 cores on TianHe 1A [19, 20], 99 s using 16,384 cores on Tianhe 2 [21,22,23,24] and 64 s using 65,536 cores on Mira [17, 25]. By improving its most time-consuming steps, such as input parallelization, kmer graph construction, and graph simplification, SWAP-Assembler 2 can scale to more than ten thousand of cores when assembling 4 terabyte genomic data [26].
In addition to gene assembly, gene identification/prediction is also very important for downstream analysis. Gene prediction attempts to identify a biological pattern in DNA sequences and predict the start and stop region of genes in the DNA sequence, or the location of protein coding regions. In eukaryotes, gene prediction and annotation are complicated due to the varying sizes of introns located between exons. Since proteins play all essential functions in the cellular environment, predicting/identifying genes that code the functional proteins in a sample is an important task. Considering the importance of gene identification, software like GENMARK [27], Glimmer [28] and Prodigal [29] respectively have been developed. These algorithms perform prediction by taking the advantage of compositional differences among coding regions, “shadow” coding regions (coding on the opposite DNA strand), and noncoding DNA. In gene prediction, there will be a large number of redundant sequences. Hobohm and Sander [30, 31] developed clustering algorithms for non-redundant gene sequences. The basic idea is to divide the gene sequence set into several classes, and then find a representative sequence for each class, and ultimately the set formed by these representative classes is the non-redundant reference gene set. The software for de-redundancy of biological gene data mainly includes NRDB90 [32], CD-HIT [33,34,35], PICSES [36], etc.
Analyzing metagenomic data has two challenges. One is the relatively low abundant species in the metagenomic samples without reference genome which leads to failure in identification of genes, and the other is the large size of the datasets. Considering the rapid increase of huge data generated by NGS technology, we believe that the combining of big data technology and distributed gene assembly software can lead to assemble and identify genes accurately. By combining many samples together, the genomic information of low abundant species in the samples has been amplified, and thus better genome assembly results and more genes can be obtained. As a result, it is possible to discover more new genes.
Considering the above facts, based on our previously developed highly scalable gene assembly software SWAP-Assembler 2, we present a workflow, WFswap, which can assemble large genomic data based on many samples and more genes in the samples can be identified/predicted. The workflow has several steps including quality control, genome assembly, gene prediction etc. A similar workflow WFsoap that relies on SOAPdenovo2 for gene assembly is also used in the paper. The test experiments show that the proposed workflow WFswap achieved better performance both for N50 and the number of identified genes compared with WFsoap. WFswap is able to identify 94.2% of the benchmark genes when tested on the 19 metagenomic samples that contains Bifidobacterium animalis subsp. lactis CNCM I-2494 [37].
2 Methods
The proposed workflow aims at predicting and identifying genes from the NGS samples. For many metagenomic samples, the proposed method can better predict/identify the genes in the sample, especially for low abundant species, since the genomic information of the low abundant species is amplified with many samples.
In order to improve the parallelism of the assembly algorithm and decouple the computational data dependence in the assembly algorithm, the method WFswap is proposed in this paper by using SWAP-Assembler 2 assembly software. SWAP-Assembler 2 proposed a mathematical model for assembly of highly extensible gene sequences called bidirectional multi-step graph model. Also, the “lock-computing-unlocking” mechanism is used to calculate the vertices or edges of a graph. Finally, optimization through input parallelization further reduces time usage.
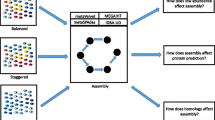
The main workflow of the model is shown in the Fig. 1. Illumina raw sequencing reads from NGS genomic samples are filtered using fastp software [38]. The reads in fastq format are then converted into fasta format by FastX. High-quality reads are then assembled together. Gene prediction is performed for the assembled gene sequences that are longer than 500 bp using MetaGeneMark [39]. After that, predicted genes from all samples were clustered resulting in the non-redundant genes. Detailed descriptions about quality control, gene assembly and prediction are as follows,

Workflow of WFswap model
-
Quality Control. There are many errors in Illumina raw sequencing reads. In order to filter these errors for a high quality control, raw sequencing reads were filtered with a quality cutoff of 20 and reads shorter than 30 bp were discarded using fastp software. In WFswap, N base in reads would be dropped during format conversion using FastX.
-
Gene Assembly. After quality control, reads were assembled into contigs with SWAP-Assembler 2 and SOAPdenovo2, which are all based on de Bruijn graph technology. Assembly parameter K-mer size was all set to 31. We dropped K-mers whose frequency is less than or equal to 1 for the purpose of reducing error information and improving assembly accuracy.
-
Gene Prediction. Gene prediction attempts to identify a biological pattern in DNA sequences and predict the start and stop regions of genes in the DNA sequence, or the location of protein coding regions. In our workflow, genes were predicted from contigs longer than 500 bp using MetaGeneMark. Any two genes with greater than 98% identity and covering more than 90% of the shorter gene were clustered together. Finally, cluster representatives shorter than 100 bp were discarded resulting in the non-redundant genes.
Different assembly algorithms used in the workflow will generate different gene assembly and prediction results. In this paper we mainly use two assembly algorithms, SWAP-Assembler and SOAPdenovo2. SOAPdenovo2 is a popular assembly algorithm based on single computing node and thus is not able to assembly large datasets, while SWAP-Assembler 2 is a highly scalable/parallel assembly tool aiming at assembling genomic data of Terabytes. The workflow with SWAP-Assembler is referred as WFswap and the workflow with SOAPdenovo2 is referred as WFsoap in this paper.
2.1 Data Sets Description
Our experiments were based on human gut microbiome samples downloaded from the website of EBI [40]. All samples were subject to Illumina deep sequencing resulting in 4.5 Gb sequence per sample on average, and a total of 23.2 billion high-quality sequencing reads with an average length of 77 bp. Among all 396 samples, nineteen of the individuals consumed a defined fermented milk product containing the previously sequenced Bifidobacterium animalis subsp. lactis CNCM I-2494, and we used this species as a benchmark to assess the ability of our method to identify the benchmark genes. On average, only 0.3% of the sequence reads in the 19 samples originated from B. animalis.
2.2 Experiment Description
In order to evaluate the results, we take the following three metrics, N50 of contigs (referred as N50), the total number of predicted genes by MetaGeneMark and the total number of predicted genes in benchmark. In this paper, we evaluate the performance of our workflows in four steps.
In the first step, we test the workflow on 30 randomly selected single samples. Each sample is subject to the workflow and the results of WFswap and WFsoap are compared. In the second step, a total of four runs were conducted. Each run contains four samples from the 19 benchmark samples and each sample is selected by once. The third step contains three experiments with different sample sizes. We use three groups of samples, group 1 with 4 samples, group 2 with 8 samples and group 3 with 12 samples. Group 2 contains all the 4 samples of group 1, group 3 contains all the samples of group 1 and group 2. In the last experiment we use 6 groups to evaluate the performance of WFswap, with sample sizes ranging from 4 to 30. The aim is to study the effect of increasing number of samples on the results of gene assembly and prediction.
3 Results and Discussion
3.1 Results for Single Samples
The gene assembly and identification results of 30 samples randomly selected from the 396 samples are shown in Table 1. These samples were subject to the two different workflow analysis, WFsoap and WFswap respectively. In order to evaluate the results, we consider the following two metrics, N50 of contigs (referred as N50), and the total number of predicted genes by MetaGeneMark. Table 1 shows the difference in N50 and the difference in the total number of predicted genes between these two workflows. A positive value in Delta_N50 and Delta_NG indicates that WFswap performs better than WFsoap. We observe that the difference in N50 ranges from 42 to 268, and for all the 30 randomly selected samples, N50 of WFswap is generally better than WFsoap.
For the non-redundant predicted genes by MetaGeneMark, an average of 2500 more genes are identified/predicted by WFswap compared with those of WFsoap. This clearly indicates the better performance of WFswap on randomly selected samples. On the other hand, 4 out of 30 samples showed negative results, indicating a poor performance for these samples by WFswap.
3.2 Results for Four Combined Samples
In this section, we assemble the combined dataset of four samples in one run using WFswap and WFsoap. For each run, we selected four samples from the 19 benchmark samples of B. animalis and every sample can only be selected once. These 19 benchmark samples consumed a defined fermented milk product containing the previously sequenced Bifidobacterium animalis subsp. lactis CNCM I-2494. Overall a total of four runs were conducted, indicated by run1, run2, run3, run4 in Table 2 which shows the assembly and gene prediction results for both WFswap and WFsoap.
In addition to the metrics used in the previous section, we also evaluate results from the perspective of the total number of benchmark genes identified by the method. MetaGeneMark is used for predicting the gene catalogs from the assembly result and each benchmark gene in the Bifidobacterium animalis subsp. lactis CNCM I-2494 is mapped to the gene catalogs using BLAST [41]. We downloaded all genes of Bifidobacterium animalis subsp. lactis CNCM I-2494 and the total number is 1658. Finally, the identified benchmark genes can be calculated from the BLAST results.
BLAST is used for searching similar sequences during which default parameter were used other than the expected value set to 0.01. The expected value is a parameter that describes the number of hits one can expect to see by chance when searching a database of a particular size. It decreases exponentially as the score of the match increases. By BLAST comparison, we mapped each gene in the benchmark set to the gene catalogs predicted from MetaGeneMark and calculated the number of predicted benchmark genes (Delta_NBG).
Table 2 shows the results for 4 combined samples, including the difference in N50 (Delta_N50), the difference in the number of predicted genes (Delta_NG) and the difference in the number of predicted benchmark genes (Delta_NBG) between these two workflows. Our observations reveal WFswap performed better in terms of Delta_N50, Delta_NG, and Delta_NBG. On average, we found 60,000 more genes with WFswap, which corresponds to about 20% of the total number of predicted genes for WFsoap. Because some samples contain a very low amount of benchmark genes, the number of genes predicted is quite different. Despite this, we still found more mapped genes with WFswap. It should be noted that in run 3, only 64 benchmark genes were identified, much less than other runs. This is because two samples used in this run have much less genomic sequence information from the benchmark gene.
3.3 Results of Concatenated and Combined Datasets
Due to the larger memory consumption, some assembly algorithms cannot perform assembly and identification on the combined dataset (all samples assembled together which is referred as COMBINE mode in the paper). One possible strategy is to assemble each sample separately and then concatenate the results together (this is referred as CAT mode in the paper). For SWAP-Assembler, more samples can be assembled together in one run since it is highly parallel and more distributed memory can be used. Since WFsoap is not be able to analyze more than 4 combined samples, it is not tested in this section.
When assembling many samples, we need to compare the difference between the two modes, CAT and COMBINE. In this section, we use three groups of samples, group one with 4 samples, group two with 8 samples and group three with 12 samples. Group 2 contains all the 4 samples of group 1 and Group 3 contains all the samples of group 1 and group 2. Each of the groups are subject to the WFswap analysis, both in CAT and COMBINE modes.
Table 3 shows the detailed N50 values, total number of predicted genes and total number of identified benchmark genes. Generally, COMBINE mode performed better in all metrics. With the increasing number of samples used in each group, more and more genes have been predicted by MetaGeneMark and similar trend is observed for the total number of identified benchmark genes, which is true for both the CAT and COMBINE modes. The reason that COMBINE mode predicted more genes is that gene assembly with COMBINE mode is able to build the de Bruijn graph for all the low abundant genomic information at once, and thus more contigs and genes can be recovered.
3.4 Results of Many Samples
From the previous analyses, we found more genes are predicted by WFswap for many samples. We want to further test the results of gene assembly and identification with even more samples. These runs are subjected to COMBINE mode since WFswap is able to assemble large dataset in one run.
We have started with 4 samples, for which WFswap was applied and the results of total numbers of predicted genes and benchmark genes are shown in Table 4. We also add 4 more samples to generate an 8 samples dataset. Similarly, datasets with 12 samples, 16 samples, 19 samples and 30 samples are generated. Each of a bigger dataset contains the samples of the smaller datasets. We performed six experiments in total and all of them are subject to the analysis in COMBINE mode. It should be noted that the first 19 samples correspond to the samples that consumed a defined fermented milk product containing the previously sequenced Bifidobacterium animalis subsp. lactis CNCM I-2494.
Table 4 shows the prediction for different samples by WFswap. As the number of samples increases, we were able to predict more genes. In terms of total number of genes in benchmark, the overall trend is also increasing. The total number of identified benchmark genes reached maximum of 1562 for 19 samples, corresponding to about 94.2% benchmark genes. This shows that many sample based method can help with gene assembly and prediction. However, for 30 samples, the total number of benchmark genes are reduced by 5 compared with that of 19 samples, this is because the added 11 samples don’t contain Bifidobacterium animalis subsp. lactis CNCM I-2494 and noise is introduced to the combined dataset.
4 Conclusion
In this article, we have presented a workflow for efficient gene assembly and identification based on many genome samples. The presented workflow is systematically tested on many samples and found that performed better for gene assembly and identification. Also, we have shown combining multiple samples together makes the gene assembly and identification of low abundant species possible. The proposed workflow WFswap is able to analyze many samples in one run, and thus the de Bruijn graph built during the simulation contains more sequence information of the low abundant species than a run with less samples. The test on the 19 benchmark samples shows WFswap is able to identify 94.2% of the benchmark genes, thus the workflow can be effectively applied to large genomic projects with terabytes of data. In future, the presented workflow can be improved to identify more genes by analyzing the bottlenecks in the method.
References
Qin, J., Li, R., Raes, J., et al.: A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010)
Warren, R.L., Sutton, G.G., Jones, S.J.M., Holt, R.A.: Assembling millions of short DNA sequences using SSAKE. Bioinformatics 4, 500–501 (2007)
Jeck, W.R., Reinhardt, J.A., Baltrus, D.A., et al.: Extending assembly of short DNA sequences to handle error. Bioinformatics 23, 2942–2944 (2007)
Dohm, J.C., Lottaz, C., Borodina, T., Himmelbauer, H.: SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing. Genome Res. 17, 1697–1706 (2007)
Bang-Jensen, G., Gutin, A., Yeo, A.: When the greedy algorithm fails. Discrete Optim. 1, 121–127 (2004)
Batzoglou, S., Jaffe, D.B., Stanley, K., et al.: ARACHNE: a whole-genome shotgun assembler. Genome Res. 12, 177–189 (2002)
Myers, E.W., Sutton, G.G., Delcher, A.L., et al.: A whole-genome assembly of Drosophila. Science 287, 2196–2204 (2000)
Huang, X., Madan, A.: CAP3: a DNA sequence assembly program. Genome Res. 9, 868–877 (1999)
Huang, X., Yang, S.P.: Generating a genome assembly with PCAP. Curr. Protoc. Bioinformatics 11 (2005). Unit11.3
de la Bastide, M., McCombie, W.R.: Assembling genomic DNA sequences with PHRAP. Curr. Protoc. Bioinformatics 11 (2007). Unit11.4
Mullikin, J.C., Ning, Z.: The phusion assembler. Genome Res. 13, 81–90 (2003)
Marcel Margulies, M.E., William, E.A., Said, A., et al.: Genome sequencing in open microfabricated high density picoliter reactors. Nature 437, 376–380 (2005)
Pevzner, P.A., Tang, H., Waterman, M.S.: An eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. 98, 9748–9753 (2001)
Luo, R., et al.: SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18 (2012)
Peng, Y., Leung, H.C.M., Yiu, S.M., et al.: IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012)
Li, D., Liu, C., Luo, R., et al.: MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015)
Meng, J., Seo, S., Balaji, P., et al.: SWAP-Assembler 2: optimization of de novo genome assembler at extreme scale. In: International Conference on Parallel Processing (2016)
Li, G., Ma, L., Song, C., et al.: The YH database: the first Asian diploid genome database. Nucleic Acids Res. 37, D1025–D1028 (2009)
Meng, J., Wang, B., Wei, Y., et al.: SWAP-Assembler: scalable and efficient genome assembly towards thousands of cores. BMC Bioinformatics 15(Suppl 9), S2–S2 (2014)
Yang, X.J., Liao, X.K., Lu, K., et al.: The TianHe-1A supercomputer: its hardware and software. J. Comput. Sci. Technol. 26(3), 344–351 (2011)
Meng, J., Wei, Y., Seo, S., et al.: SWAP-Assembler 2: scalable genome assembler towards millions of cores – practice and experience. In: IEEE/ACM International Symposium on Cluster (2015)
Liao, X., Xiao, L., Yang, C., Lu, Y.: MilkyWay-2 supercomputer: system and application. Front. Comput. Sci. 8, 345–356 (2014)
Xu, W., Lu, Y., Li, Q., et al.: Hybrid hierarchy storage system in MilkyWay-2 supercomputer. Front. Comput. Sci. 8, 367–377 (2014)
Liao, X., Pang, Z., Wang, K., et al.: High performance interconnect network for Tianhe system. J. Comput. Sci. Technol. 30, 259–272 (2015)
Kumaran, K.: Introduction to Mira, in Code for Q Workshop (2016)
Meng, J., Guo, N., Ge, J., et al.: Scalable assembly for massive genomic graphs. In: Proceedings of the 17th IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing (CCGRID 2017), Madrid (2017)
Borodovsky, M., Mclninch, J.: GeneMark: parallel gene recognition for both DNA strands. Comput. Chem. 17, 123–133 (1993)
Salzberg, S., Delcher, A., Kasif, S., White, O.: Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 26, 544–548 (1998)
Hyatt, D., Chen, G.L., Locascio, C.L., et al.: Prodigal: prokaryotic gene recognition and transla-tion initiation site identification. BMC Bioinformatics 11, 119 (2010)
Hobohm, U., Scharf, M., Schneider, R., et al.: Selection of representative protein data sets. Protein Sci. 1(3), 409–417 (2010)
Hobohm, U., Sander, C.: Enlarged representative set of protein structures. Protein Sci. 3(3), 522–524 (2010)
Holm, L., Sander, C.: Removing near-neighbour redundancy from large protein sequence collections. Bioinformatics 14(5), 423–429 (1998)
Li, W., Jaroszewski, L., Godzik, A.: Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 17(3), 282–283 (2001)
Li, W., Jaroszewski, L., Godzik, A.: Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18(1), 77–82 (2002)
Li, W.: Fast program for clustering and comparing large sets of protein or nucleotide sequences. In: Nelson, K.E. (ed.) Encyclopedia of Metagenomics. Springer, Boston (2015). https://doi.org/10.1007/978-1-4899-7478-5
Wang, G., Dunbrack Jr., R.L.: PISCES: a protein sequence culling server. Bioinformatics 19(12), 1589 (2003)
Chervaux, C., Grimaldi, C., Bolotin, A., et al.: Genome sequence of the probiotic strain Bifidobacterium animalis subsp. lactis CNCM I-2494. J. Bacteriol. 93(19), 5560–5561 (2011)
Shifu, C., Yanqing, Z., Yaru, C., et al.: fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018)
Zhu, W., Lomsadze, A., Borodovsky, M.: Ab initio gene identification in meta-genomic sequences. Nucleic Acids Res. 38, e132 (2010)
Park, Y.M., Squizzato, S., et al.: The EBI search engine: EBI search as a service – making biological data accessible for all. Nucleic Acids Res. 45, W545–W549 (2017)
Altschul, S., Warren, G., Miller, W., Eugene, M., Lipman, D.: Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990)
Acknowledgements
This work was supported by National Key Research & Development Program of China under grant No. 2016YFB0201305, Shenzhen Basic Research Fund under grant no. JCYJ20160331190123578, JCYJ20170413093358429 & GGFW2017073114031767, National Science Foundation under grant no. 61702494 and U1813203. We would also like to thank the funding support by the Shenzhen Discipline Construction Project for Urban Computing and Data Intelligence, Youth Innovation Promotion Association, CAS to Yanjie Wei.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Zheng, Z., Guo, N., Saravanan, K.M., Wei, Y. (2019). Efficient Gene Assembly and Identification for Many Genome Samples. In: Xu, R., Wang, J., Zhang, LJ. (eds) Cognitive Computing – ICCC 2019. ICCC 2019. Lecture Notes in Computer Science(), vol 11518. Springer, Cham. https://doi.org/10.1007/978-3-030-23407-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-23407-2_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-23406-5
Online ISBN: 978-3-030-23407-2
eBook Packages: Computer ScienceComputer Science (R0)