
Abstract
Clinical observations show that human microorganisms get involved in various human biological processes. The disruption of a symbiotic balance for host-microbiota relationship is found to cause different types of human complex diseases. Discoverying the associations between microbes and the host health statuses that they affect could provide great insights into understanding the mechanisms of diseases caused by microbes. However, experimental approaches are time-consuming and expensive. Little effort has been done to develop computational models for predicting pathogenic microbes on a large scale. The prediction results yielded by such models are anticipated to boost the identification and characterization of potential human pathogenic microbes. Based on the assumption that microbes of similar characters tend to get involved in diseases of similar symptoms forming functional clusters, in this paper, we develop a group based computational model of Bayesian disease-oriented ranking for inferring the most potential microbes associated with human diseases. It is the first attempt to predict this kind of associations by using 16S rRNA gene sequences. Based on the sequence information of genes, we use two computational approaches (BLAST+ and MEGA 7) to measure how similar each pairs of microbes are from different aspects. On the other hand, the similarity of diseases is computed based on MeSH descriptors. Using the data collected from HMDAD database, the proposed model achieved AUCs of 0.9456, 0.8266, 0.8866 and 0.8926 in leave-one-out, 2-fold, 5-fold and 10-fold cross validations, respectively. Besides, we conducted a case study on colorectal carcinoma and found that 16 out of top-20 predicted microbes can be confirmed by the published literatures. The prediction result is publicly released and anticipated to help researchers to preferentially validate these promising pathogenic microbe candidates via biological experiments.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others

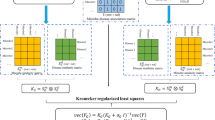
References
Clemente, J.C., Ursell, L.K., Parfrey, L.W., Knight, R.: The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012)
Sender, R., Fuchs, S., Milo, R.: Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340 (2016)
Savitz, L.D.: The human microbiota: the role of microbial communities in health and disease. Acta Biol. Colomb. 21, 5–15 (2016)
Donia, M.S., et al.: A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158, 1402–1414 (2014)
Davenport, E.R., Mizrahi-Man, O., Michelini, K., Barreiro, L.B., Ober, C., Gilad, Y.: Seasonal variation in human gut microbiome composition. PLoS One 9, e90731 (2014)
Mason, M.R., Preshaw, P.M., Nagaraja, H.N., Dabdoub, S.M., Rahman, A., Kumar, P.S.: The subgingival microbiome of clinically healthy current and never smokers. ISME J. 9, 268–272 (2015)
Manichanh, C., et al.: Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211 (2006)
Thibault, R., Blachier, F., Darcy-Vrillon, B., de Coppet, P., Bourreille, A., Segain, J.P.: Butyrate utilization by the colonic mucosa in inflammatory bowel diseases: a transport deficiency. Inflamm. Bowel Dis. 16, 684–695 (2010)
Huang, Z.A., Wen, Z., Deng, Q., Chu, Y., Sun, Y., Zhu, Z.: LW-FQZip 2: a parallelized reference-based compression of FASTQ files. BMC Bioinform. 18, 179 (2017)
Hartman, A.L., Riddle, S., McPhillips, T., Ludascher, B., Eisen, J.A.: Introducing W.A.T.E.R.S.: a workflow for the alignment, taxonomy, and ecology of ribosomal sequences. BMC Bioinform. 11, 317 (2010)
Ma, W., et al.: An analysis of human microbe-disease associations. Brief. Bioinform. 18, 85–97 (2017)
You, Z.H., et al.: PBMDA: a novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 13, e1005455 (2017)
Mork, S., Pletscher-Frankild, S., Palleja Caro, A., Gorodkin, J., Jensen, L.J.: Protein-driven inference of miRNA-disease associations. Bioinformatics 30, 392–397 (2014)
Huang, Y.A., You, Z.H., Gao, X., Wong, L., Wang, L.: Using weighted sparse representation model combined with discrete cosine transformation to predict protein-protein interactions from protein sequence. Biomed. Res. Int. 2015, 902198 (2015)
Huang, Y.A., You, Z.H., Chen, X., Yan, G.Y.: Improved protein-protein interactions prediction via weighted sparse representation model combining continuous wavelet descriptor and PseAA composition. BMC Syst. Biol. 10, 120 (2016)
Y.A. Huang, Z.H. You, X. Chen: A systematic prediction of drug-target interactions using molecular fingerprints and protein sequences. Curr. Protein Peptide Sci. (2016)
Coenye, T., Vandamme, P.: Intragenomic heterogeneity between multiple 16S ribosomal RNA operons in sequenced bacterial genomes. FEMS Microbiol. Lett. 228, 45–49 (2003)
Chen, X., Huang, Y.A., You, Z.H., Yan, G.Y., Wang, X.S.: A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 33, 733–739 (2017)
Huang, Z.A., et al.: PBHMDA: path-based human microbe-disease association prediction. Front. Microbiol. 8, 233 (2017)
Katz, L.: A new status index derived from sociometric analysis. Psychometrika 18, 39–43 (1953)
Pruitt, K.D., Tatusova, T., Maglott, D.R.: NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65 (2007)
Lipscomb, C.E.: Medical subject headings (MeSH). Bull. Med. Libr. Assoc. 88, 265–266 (2000)
Wang, D., Wang, J., Lu, M., Song, F., Cui, Q.: Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 26, 1644–1650 (2010). (Oxford, England)
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., Madden, T.L.: NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9 (2008)
Camacho, C., et al.: BLAST+: architecture and applications. BMC Bioinform. 10, 421 (2009)
Kumar, S., Stecher, G., Tamura, K.: MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016)
Larkin, M.A., et al.: Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007)
Thomas, R.H.: Molecular evolution and phylogenetics. Heredity 86, 385 (2001)
Hu, Y., Koren, Y., Volinsky, C.: Collaborative filtering for implicit feedback datasets. In: Eighth IEEE International Conference on Data Mining, pp. 263–272 (2009)
Pan, R., et al.: One-class collaborative filtering. In: Eighth IEEE International Conference on Data Mining, pp. 502–511 (2008)
Rendle, S., Freudenthaler, C., Gantner, Z., Schmidt-Thieme, L.: BPR: Bayesian personalized ranking from implicit feedback. In: Conference on Uncertainty in Artificial Intelligence, pp. 452–461 (2009)
Pan, W., Chen, L.: GBPR: group preference based Bayesian personalized ranking for one-class collaborative filtering. In: International Joint Conference on Artificial Intelligence, pp. 2691–2697 (2013)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Huang, YA. et al. (2019). Precise Prediction of Pathogenic Microorganisms Using 16S rRNA Gene Sequences. In: Huang, DS., Jo, KH., Huang, ZK. (eds) Intelligent Computing Theories and Application. ICIC 2019. Lecture Notes in Computer Science(), vol 11644. Springer, Cham. https://doi.org/10.1007/978-3-030-26969-2_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-26969-2_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-26968-5
Online ISBN: 978-3-030-26969-2
eBook Packages: Computer ScienceComputer Science (R0)