
Abstract
In the last few years, Deep Learning (DL) gained more and more impact on drug design because it allows a huge increase of the prediction accuracy in many stages of such a complex process. In this paper a Virtual Screening (VS) procedure based on Convolutional Neural Networks (CNN) is presented, that is aimed at classifying a set of candidate compounds as regards their biological activity on a particular target protein. The model has been trained on a dataset of active/inactive compounds with respect to the Cyclin-Dependent Kinase 1 (CDK1) a very important protein family, which is heavily involved in regulating the cell cycle. One qualifying point of the proposed approach is the use of molecular fingerprints as a suitable embedding for describing molecules; up to our knowledge there is no Deep Learning approach for VS that makes use of such descriptor. Several kinds of fingerprints are reported in the scientific literature to address different aspects of both the structure and the local properties of a molecule. Both 1D and 2D CNNs have been trained to test the performance of each single descriptor separately, along with suitable ensembles of multiple descriptors for the same compound; the best performing architecture has been used for prediction. The CNN architectures are described in detail, and the results are compared with some recent approaches for Virtual Screening with respect to Cyclin-Dependent Kinase proteins that do not use molecular fingerprints as their descriptor.
Supported by PON “Ricerca e Innovazione” 2014–2020, Azione 1.1: Dottorati innovativi con caratterizzazione industriale.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others
Keywords
1 Introduction
Drug discovery is the very long and complex process leading to the development of a new medication, where several steps and loops are involved. Indeed, one of the most relevant parts in the drug discovery cycle is drug design when one already knows the biological target the new compound has to bind to. In general, a biological target is an enzyme or a protein. In a modern drug design setup, many compounds are screened to assess the best matching ones as regards their ability of either inhibiting or activating the target associated to a particular disease. Such a process is also referred to as Inverse Pharmacology or Target-based Drug Design.
As part of his/her work, the drug designer needs to consult large public or private databases to retrieve information about existing molecules. Such queries can have different nature; particularly, it is necessary to investigate how two molecules can bind with each other according to purely chemical criteria, taking into account biological constraints due to the molecule toxicity for the human organism. From the data point of view, these queries return very heterogeneous information ranging from a whole molecule graph representation to textual and numerical data.
In recent years, computer aided drug design has gained increasing importance to speed up the whole process thus reducing the cost of developing new drugs significantly. Virtual Screening (VS) refers exactly to an automated procedure aimed at selecting those molecules that are likely to be active on the desired target. Such procedures range from similarity search to Machine Learning (ML) approaches, and all of them rely on using suitable numerical descriptors of both the structure and some chemical properties of the candidate compound.
Virtual Screening can be regarded as a classification task in the ML perspective; typical approaches are Support Vector Machines and Random Forests. In the Deep Learning era, both Convolutional and Recurrent Neural Networks are used for VS and in other fields of Pharmacology like the prediction of chemical reactions. The reader is referred to [8] for a recent and thorough review.
In this paper a VS procedure based on Convolutional Neural Networks (CNN) is presented, that is aimed at classifying a set of candidate compounds as regards their biological activity on the Cyclin-Dependent Kinase 1 (CDK1) a very important protein family, which is heavily involved in regulating the cell cycle. This work is part of a more wide research oriented to the development of new CDK1 inhibitors starting from natural products, to be used in cancer therapy. The choice of this target is given by the previous experience of the research group in the CDK1 modulators and the fact that canonical VS approaches on CDKs do not respond properly to activity prediction because of high structural similarity between different kinases binding sites. The importance of the target is given by its validation as drug target. It is an archetypal kinase acting as central regulator that drives cells through G2 phase and mitosis. Its importance in tumorigenesis has been demonstrated by the evidence that, unlike other CDKs, loss of CDK1 in the liver confers complete resistance against tumor formation demonstrating its role in the cancer development [4].
The proposed CNN architecture makes use of molecular fingerprints as the numerical descriptor of each candidate molecule, and this is quite a novelty as regards DL approaches for biological activity prediction. Several kinds of fingerprints are reported in the scientific literature to address different aspects of both the structure, and the local properties of a molecule. The same fingerprint can be devised also with different sizes. As a consequence, experiments have been performed to test the performance of each single descriptor separately by training 1D CNNs. Also 2D CNNs have been trained on suitable combinations of different equal sized fingerprints for the same compound, to take into account all the diverse information pieces coming from such descriptors at once. As it was expected, multiple fingerprints performed better, and results are reported as regards both the best ensemble and the best size. Moreover, we compared our results with some recent approaches for Virtual Screening with respect to Cyclin-Dependent Kinase proteins that do not use molecular fingerprints as their descriptor.
The rest of the paper is arranged as follows. Section 2 reports a review of the state of te art in ML and DL in drug design, and particularly in VS. Section 3 contains a description of molecular fingerprints along with the details on the datasets, and the CNN architectures. Section 4 contains results and comparisons, while conclusions are drawn in Sect. 5.
2 State of the Art
Clinical candidate molecules selected by drug detection must have a profile responding to different criteria, that are based not only on the effect potency but also on the selectivity, safety as well as the so called ADMET properties (Absorption, Distribution, Metabolism, Excretion and Toxicity). Therefore, the design of the optimal compound is a multidimensional challenge involving different aspects of Chemistry and Biology, which is faced using ML. One key aspect for ML approaches gaining success in property prediction is the possibility to access and mining large data sets that contain heterogeneous information. Until recent years, the best performing ML techniques were “shallow” ones [9] that is support vector machines (SVM) and decision trees, particularly ensemble methods like Random Forest (RF). All these ML models should be iteratively refined with new experimental data to increase model reliability and predictive power.
The Kinase protein family presents a huge variety, and contains a very high number of proteins so it provides an amount of data that is well suited for ML approaches oriented to VS for novel kinase inhibitors. In [11] Bayesian models were generated for building Quantitative Structure-Activity Relationship (QSAR) models on different kinases from a large, but sparsely populated data matrix of more than 100,000 compounds. Random Forest has been applied in another case study for predicting kinase activities on hundreds of kinases starting from publicly available data sets integrated with in-house data [12]. In several examples, Random Forest models showed a higher reliability in prediction when compared to other approaches, but they perform worse than deep neural networks.
DNNs have been used for predicting different properties such as biological activity, ADMET properties, and physicochemical parameters demonstrating reliable and robust predictivity capabilities with high sensitivity when used on different targets [7, 15]. AtomNet was one of the first CNNs designed for drug discovery to predict bio-activity of small molecules [16]. CNNs have been used also to predict several properties such as the kinetic energy of hydrocarbons as a function of electron density [17]. Several DNN architectures use Simplified Molecular Input Line Entry System (SMILES) as their input data [3, 6, 14]. SMILES is actually a simple chemical language whose rules allow building string descriptors that can represent both molecular structures and reactions.
The architecture presented in this work relies on CNNs and molecular fingerprints for VS of compounds as regards their biological activity on the CDK1 protein. Up to our knowledge there is no other approach in the literature making use of molecular fingerprints as the data embedding for training a VS deep neural model for bioactivity on CDK1. A detailed description of molecular fingerprints is reported in the next section. As regards CDK-oriented VS, Li et al. [10] propose a least-squares support vector machine (LS-SVM) trained on molecular descriptors to build a Structure-Activity Relationship (SAR) model to classify oxindole-based inhibitors of CDK1 and CDK2. In another study, Pereira et al. [13] present DeepVS a deep learning approach for docking-based virtual screening that has been tested on the data sets included in the Directory of Useful Decoys (DUD) where only CDK2 were used. The results reported in the two previous works were considered to compare the performance of the proposed architecture, computing the fingerprints of the data sets they used. The results of our experiments against DeepVS are reported in Sect. 4, while results reported by the authors of LS-SVM were not useful because they used a very small data set made by 82 compounds referred to by numerical IDs, that was extracted manually from another paper in the literature. As a consequence it was not possible for us to retrieve the original compounds from any chemical database for generating their fingerprints.
3 Materials and Methods
In this section we report a description of molecular fingerprints, the data sets used in our experiments, and the architecture of the proposed CNNs.
3.1 Molecular Fingerprints
Modern approaches in Chemoinformatics have focused on the use of ML techniques applied to fingerprints instead of classical molecular descriptors. The reason is that fingerprints contain information on chemical groups and paths; they provide complete information about molecular complexity thus allowing a more robust comparison between two or more structures than molecular descriptors do. SMILES descriptors also convey information on molecular structures but their inherent string form needs the cycles to be cut, and the description of the same molecule is not unique thus a “SMILES canonicalization” is also needed.
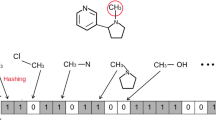
Molecular fingerprints are generated analyzing each atom along with its neighborhood till 6 or 7 bonds away. Such a neighborhood is searched for a set of predefined molecular substructures, the so called patterns, that is atom types, bond types, presence of rings, ans so on. After having enumerated all the patterns in the molecule, each of them is used as a seed for a hashing function that outputs in general 4 to 5 index positions whose corresponding bits are set to 1 in the “pattern fingerprint”; such a fingerprint is bit-wise ORed to the molecular one. Actually the hashing function can cause a bit collision so we are not guaranteed of the effective presence of a particular pattern unless at least one of its bits is unique. On the other hand, a molecular substructure is absent if all its bits are set to 0 in the fingerprint. A simplified fingerprint generation procedure is reported in Fig. 1.

Simplified fingerprint generation: the hashing function sets just 1 bit per pattern
Molecular fingerprints are generated using different approaches as regards both the neighborhood definition and the size. In our experiments we tested six among the most popular fingerprints: RDKit, Morgan, AtomPair, Topological Torsion, Layered, and FeatMorgan. All of them differ in the choice of the paths along the molecule to devise patterns, and particularly the Morgan and FeatMorgan fingerprint are circular that is they generate patterns by going through each atom of the molecule and obtaining all possible paths through this atom with a specific radius. Each unique path is then hashed into the fingerprint: the larger the radius, the larger fragments are encoded. Fingerprints’ length can range from 256 to 4K bits; in classical VS, different size fingerprints are compared by “folding” them. The two halves of the longest fingerprint are bit wise OR-ed thus obtaining a new fingerprint whose length is one half of the original one. In our experiments, we tested each kind of fingerprints using three different lengths: 256, 512, and 1024 bits, while the radius of the circular fingerprints was set to 2, that is the conventional value.
From the computational point of view, VS procedures take advantage from the fact that fingerprints are not too sparse bit vectors. Non ML approaches perform different search strategies where some well known similarity measures are used like Tanimoto, Cosine, Dice, Euclid, or Twersky; such measures are computed starting from the number of 1s counted respectively in each fingerprint, and on the number of 1s in common between the two fingerprints.
Fingerprints have been also learnt from molecular graphs using CNNs as reported in [5]. In this work, a single convolutional layer with softmax activation is used in place of the hashing function to produce the bits indexing of a atom neighborhood collected in the same way as circular fingerprints do. Authors report very good performance in predicting both solubility and toxicity from two purposely defined data sets, but the approach suffers from a high computational cost when compared with direct use of circular fingerprints.
3.2 Data Sets
The data used in our experiments where extracted from the well known CheMBL molecular database [1]. Biological activity of the tested compounds was measured using the half maximal inhibitory concentration parameter (\(IC_{50}\)) that is the amount of substance which is needed to inihibit the target protein (i.e. CDK1) by one half. A molecule has been considered active when \(IC_{50}\,\le \,9\ \mathrm {\mu M}\), otherwise it is inactive.
Data preparation was accomplished using the KNIME data analysis platform [2], and a workflow was implemented to prepare both the training and the test set. Activity data for 1830 compounds on the CDK1 target were taken from the CHEMBL308 ID were CDK1 is considered as a single protein, and the CHEMBL1907602 ID were it is considered as a protein complex.
At first, incomplete data were deleted; the training set was then made using 1432 samples with a perfect balancing between the two class labels. Particularly, 716 active samples and 662 inactive ones were selected from the CHEMBL308 ID, while 54 inactive samples were selected from the CHEMBL1907602 ID. The test set was made as a whole by 175 inactive molecules coming from the CHEMBL1907602 ID, and 100 active samples coming from the CHEMBL308 ID.
Data in the two CheMBL IDs were searched for duplicates that were removed to avoid repetitions in both the training and the test set. However it is worth noting that in the same data set there may be two times the same molecule with very different \(IC_{50}\) value coming from two different biological assays. We have not used data augmentation because it is not possible to generate molecular fingerprints and predict whether they are active molecules or not in a specific biological assay but we have used 5-cross validation.
3.3 The CNN Architecture
Molecular fingerprint generation acts as a transform on the molecular structure from the spatial domain to a suitable Vector Space Representation. A fingerprint represents the corresponding molecule “as a whole” that is it conveys information about the presence of a particular substructure but not on its exact position or its repetition in different sites of the same molecule. Moreover, we want to perform a binary classification between active and inactive compounds, and biological activity is mostly related to the presence/absence of particular substructures which in turn are well suited to bind to the target protein. As a consequence, a CNN architecture appeared to be the best choice to classify molecular fingerprints.
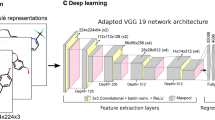
In this study we present two CNN architectures that have been trained from scratch; the first is a 1D CNN trained on single fingerprints, and a 2D network where each compound was represented by an ensemble of equal length and different kind fingerprints arranged as a bi-dimensional matrix. The second network is aimed at modeling those structural subtleties that can not be represented by any single descriptor alone. In general, different patterns are searched for in each fingerprint kind, and also the same pattern is searched in different ways. Both networks consist of 4 convolutional layers with 512, 256, 128, 64 filters respectively with ReLU activation, each followed by a 2 \(\times \) 2 Max Pooling, while they differ only in the convolutional kernel dimensions. Classification is achieved through a MLP with 1024, 512, and 256 ReLU units respectively, while the ouput is a sigmoidal unit as we want binary classification. The overall architecture is reported in Fig. 2. Hyperparameters tuning was performed as a grid search in the following sets of values; Convolutional filters tested were [1024, 512, 256, 128, 64] in combination with all Keras padding value; learning rate were multiplied by 10 in the ranges \([10^{-6},1;2\cdot 10^{-5},0.2]\). Dropout probabilities where in the range [0.2, 0.9] with step 0.1, all the available optimizers in Keras were tested. Bi-dimensional tested kernel sizes were \(\{(20,2), (20,1), (15,2), (15,1), (5,2), (5,1), (4,2), (4,1), (3,2), (3,1)\}\), while 1D tested kernels were \(\{2, 3, 4\}\). Batch sizes were doubled in the range [8, 128]. Early sopping was used to devise training epochs. Table 1 shows the best choices for all the hyperparameters. Due to the low number of samples, small size fingerprints were tested with a number of epochs greater than 55; retraining was performed with 70, 100 and 120 epochs, and the minimum loss was achieved with 100 epochs. No overfitting was encountered with this setup. Hyperparameter optimization took about 150 h to be accomplished on a GPU NVIDIA GTX1060 6 GB, 1280 CUDA Cores, while each experiment took about 20 min.

Bi-dimensional architecture of the network
4 Results and Discussion
The first set of experiments where devoted to devise the best performing fingerprint type/size in predicting biological activity, and 1D CNN was used. Table 2 reports the best test results for each fingerprint size along with its type. Here and in the following tables, best results are highlighted in bold. The table reports the achieved test accuracy, the F1-score, and the AUC value, which is used commonly when comparing two approaches in the drug design literature. Both a SVM and a Random Forest model were trained on our data sets to validate the performance of our model. The results of such experiment are reported in Fig. 3. As it was expected, ML approaches have a very poor accuracy performance (SVM = 0.9081, RF = 0.9081) if compared to ours best architecture (0.9345), despite the better AUC value shown in Fig. 3.

ROC Curves comparison of the proposed architecture with classical ML approaches; (a) best performing 1D CNN (L-512); (b) SVM; (c) Random Forest.
The second round of experiments was aimed at devising the best fingerprint combination/size for biological activity prediction using 2D CNN. The idea behind this experiments is that different fingerprints for the same molecule contain many different patterns, which in turn describe different molecular substructures. Also different sizes correspond to patterns with variable length. As a consequence, a set of fingerprints arranged as a 2D matrix can act as a better descriptor for molecular substructures than a single one can do. Table 3 reports the overall results for different fingerprint sizes.
As it is reported in Tables 2 and 3, the best performance is achieved with the set of Morgan, Topological Torsion and Layered 512 bit fingerprints (MTL-512). Layered fingerprints are always among the best performing descriptor regardless their size. Moreover, 512 Layered is exactly the best performing descriptor in the 1D CNN architecture. It is trivial to say that 512 bit is the input data size that best suits to the network capacity as it is defined by its architecture. As regards the fingerprint types, it is difficult to devise an exact explanation of the results due to the random process involved in the generation of molecular fingerprints. It is not possible to devise precise patterns in precise positions that are mainly responsible for the network performance. Anyway, we can say that Layered fingerprints have a particular hashing scheme that allows accommodating substructure information with high level of detail so it is reasonable that 1D CNN achieved its best performance using this kind of fingerprint. As regards the 2D CNN’s performance, it is worth noting that MTL-512 fingerprints together span all the diverse criteria to search for patterns so it seems quite reasonable that such a triple produced the best result.
We further validated our architecture against the DeepVS network, which is presented in [13], and deals with VS versus CDK proteins even if there are some differences with our work.
DeepVS was trained on the CDK2 protein only; the authors tested their network with a subset of the CHEMBL301 data set, which is extracted from the DUD-E data set (798 active molecules and 28,329 decoys). At first, the entire CHEMBL301 data set that consists of 1528 compounds (956 CDK2-active molecules, and 572 inactive ones) was used to test the MTL-512 2D CNN. In this experiment our network achieved AUC = 0.8030 that is a very good result when compared with AUC = 0.82 achieved by DeepVS, which was trained purposely for CDK2. As some compounds in CHEMBL301 are also active on CDK1, we removed explicitly all of them to stress the network performance. As a result, we obtained AUC = 0.678, which shows an obvious decrease; this still remains a satisfactory result if related to human performances in VS, and also classical ML approaches.
5 Conclusions
A novel CNN architecture has been presented in this work, that is trained on the molecular fingerprints to predict biological activity of candidate medical compounds versus the CDK1 protein target. The main novelty of the paper relies on performing Deep Learning based VS starting from molecular fingerprints for CDK1 that is a very important biological target for its direct implication in the etiology of various cancerous forms. One qualifying point of our approach is that fingerprints capture molecular structures according to different criteria and are already accepted as molecular descriptors by the chemoinformatics society. Another novelty of the approach is the use of fingerprint matrices, in order to keep direct information from single fingerprint and indirect information from the combination of the same. Their shape already makes them an embedding that lends itself perfectly to the intended use. Fine tuning of hyperparameters has been carried on along with several experiments with different fingerprint types and sizes. Early results are satisfactory, and indicate that VS of suitable arrangements of multiple fingerprint types, each addressing different ways of representing molecular substructures, performs better than a single fingerprint approach. Our architecture has been also compared to both classical ML and other state-of-the art DL approaches even if trained on different data, and for a different task. Future work will be oriented to a deep understanding of the relation between particular fingerprints and biological activity prediction, and to build a general architecture for screening compounds with respect to all the CDK family.
References
CHeMBL Database. https://www.ebi.ac.uk/chembl/. Accessed 24 Sept 2018
Berthold, M.R., et al.: KNIME - the konstanz information miner: version 2.0 and beyond. SIGKDD Explor. Newsl. 11(1), 26–31 (2009)
Bjerrum, E.J.: SMILES enumeration as data augmentation for neural network modeling of molecules. CoRR, abs/1703.07076 (2017)
Diril, M.K., et al.: Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Nat. Acad. Sci. U.S.A. 109(10), 3826–3831 (2012)
Duvenaud, D.K. et al.: Convolutional networks on graphs for learning molecular fingerprints. In: Advances in Neural Information Processing Systems, pp. 2224–2232 (2015)
Fooshee, D., et al.: Deep learning for chemical reaction prediction. Mol. Syst. Des. Eng. 3, 442–452 (2018)
Ghasemi, F., Mehridehnavi, A., Pérez-Garrido, A., Pérez-Sánchez, H.: Neural network and deep-learning algorithms used in QSAR studies: merits and drawbacks. Drug Discovery Today 23(10), 1784–1790 (2018)
Jing, Y., Bian, Y., Hu, Z., Wang, L., Xie, X.-Q.S.: Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the big data era. AAPS J. 20, 58 (2018)
Lavecchia, A.: Machine-learning approaches in drug discovery: methods and applications. Drug Discovery Today 20(3), 318–331 (2015)
Li, J., Liu, H., Yao, X., Liu, M., Hu, Z., Fan, B.: Structure-activity relationship study of oxindole-based inhibitors of cyclin-dependent kinases based on least-squares support vector machines. Anal. Chim. Acta 581(2), 333–342 (2007)
Martin, E., Mukherjee, P., Sullivan, D., Jansen, J.: Profile-QSAR: a novel meta -QSAR method that combines activities across the kinase family to accurately predict affinity, selectivity, and cellular activity. J. Chem. Inf. Model. 51(8), 1942–1956 (2011)
Merget, B., Turk, S., Eid, S., Rippmann, F., Fulle, S.: Profiling prediction of kinase inhibitors: toward the virtual assay. J. Med. Chem. 60(1), 474–485 (2017)
Pereira, J.C., Caffarena, E.R., dos Santos, C.N.: Boosting docking-based virtual screening with deep learning. J. Chem. Inf. Model. 56(12), 2495–2506 (2016)
Segler, M.H.S., Kogej, T., Tyrchan, C., Waller, M.P.: Generating focused molecule libraries for drug discovery withrecurrent neural networks. ACS Central Sci. 4(1), 120–131 (2018)
Varnek, A., Baskin, I.: Machine learning methods for property prediction in chemoinformatics: Quo Vadis? J. Chem. Inf. Model. 52(6), 1413–1437 (2012)
Wallach, I., Dzamba, M., Heifets, A.: AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. CoRR, abs/1510.02855 (2015)
Yao, K., Parkhill, J.: Kinetic energy of hydrocarbons as a function of electron density and convolutional neural networks. J. Chem. Theory Comput. 12, 1139–1147 (2016)
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Mendolia, I., Contino, S., Perricone, U., Pirrone, R., Ardizzone, E. (2019). A Convolutional Neural Network for Virtual Screening of Molecular Fingerprints. In: Ricci, E., Rota Bulò, S., Snoek, C., Lanz, O., Messelodi, S., Sebe, N. (eds) Image Analysis and Processing – ICIAP 2019. ICIAP 2019. Lecture Notes in Computer Science(), vol 11751. Springer, Cham. https://doi.org/10.1007/978-3-030-30642-7_36
Download citation
DOI: https://doi.org/10.1007/978-3-030-30642-7_36
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-30641-0
Online ISBN: 978-3-030-30642-7
eBook Packages: Computer ScienceComputer Science (R0)
