
Abstract
In this paper we present a method based on genetic algorithm capable of analyzing a significant number of features obtained from fractal techniques, Haralick texture features and curvelet coefficients, as well as several selection methods and classifiers for the study and pattern recognition of colorectal cancer. The chromosomal structure was represented by four genes in order to define an individual. The steps for evaluation and selection of individuals as well as crossover and mutation were directed to provide distinctions of colorectal cancer groups with the highest accuracy rate and the smallest number of features. The tests were performed with features from histological images H&E, different values of population and iterations numbers and with the k-fold cross-validation method. The best result was provided by a population of 500 individuals and 50 iterations applying relief, random forest and 29 features (obtained mainly from the combination of percolation measures and curvelet subimages). This solution was capable of distinguishing the groups with an accuracy rate of 90.82% and an AUC equal to 0.967.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Colorectal cancer is a malignant tumour that develops on the internal wall of the intestine (colon) or rectum [2]. The main reasons for studying this disease are the number os cases and mortality. The International Agency for Research on Cancer (IARC) presented a study in which colorectal cancer was defined as the third most common cancer in men (746,000 cases) and the second most common one for women (614,000 cases). The number of mortalities was 694,000 and the highest incidence of 52% of deaths occurred in less developed regions of the world [17]. The diagnosis for colorectal cancer can be made through sigmoidoscopy or by colonoscopy. Confirmation occurs by biopsies of the tissues stained with hematoxylin and eosin (H&E) and microscopically analyzed by pathologists.
The main difficulty for a medical diagnosis is the evaluation of the severity of abnormal findings when there are different opinions between inter and intraobservers [7, 10]. This fact has motivated the development of systems known as computer-aided diagnosis (CAD) [36] to support specialists in research and decision-making. A common challenge observed in proposals of CAD systems is to indicate the best combination between the selection and classification algorithms to achieve the highest success rates using the smallest features number [10, 15]. In this context, solutions obtained from metaheuristics models were relevant for different sorts of medical images. The techniques inspired by analogies found in nature or in evolutionary processes are worth mentioning, such as the methods based on genetic algorithms (GA) for the diagnosis of esophagus cancer [28], lung cancer, brain tumors, prostate cancer and leukemia [21].
A GA is a metaheuristic widely known in the literature and its main advantage in comparison with other evolutionary strategies is to have a structure that makes it possible to represent plausible new organizational forms (individuals) from a successful previous organizational construct (crossover) [5] without losing critical information from the problem [26, 34]. Despite of the different strategies considering genetic algorithms for the study and development of CAD, such as diagnosis of cardiac diseases [3] and lung cancer [24], the models available in the literature did not explore the method in order to determine the best combination of features, selection algorithms and classifiers [13, 22] in the context of histological images and diagnosis of colorectal cancer. Therefore, in this work we present a method based on a GA capable of analyzing a significative number of features obtained from fractal techniques, Haralick texture features and curvelet coefficients, as well as selection methods and classifiers in order to indicate an acceptable solution for the diagnosis of colorectal cancer. This type of study contributes significantly to the literature focused on the theme, especially with the development and improvement of CAD systems. The main contributions of the proposal are:
-
1.
A method based on genetic algorithm capable of analyzing a significative number of features, selection methods and classifiers for the study and pattern recognition of colorectal cancer;
-
2.
An approach capable of indicating the best features in order to separate benign and malignant colorectal cancer groups;
-
3.
Information about methods and features which support development and enhancement of CAD systems.
2 Methodology
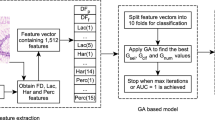
Each individual (genetic’s code bearer) was defined as a chromosome structure composed by four genes, represented by integer numbers. The information stored in each gene or genetic code defines a specific combination: (\(G_{id}\)) (the individual’s identification), selection method (\(G_{sel}\)), classification method (\(G_{clf}\)) and the number of features considered in the classification process (\(G_{num}\)). The initial values attributed to the \(G_{sel}\), \(G_{clf}\) and \(G_{num}\) genes were random. The structure described is illustrated in Fig. 1. Considering this structure, a population was defined. Each combination is unique and associated with an identifier \(G_{id}\) to define an acceptable solution.

Chromosome structure defined to represent an individual (\(G_{id}\))
It is important to emphasize that each gene \(G_{sel}\) identifies a method capable of producing a ranking of the most significant features in order to distinguish the datasets under investigation. The explored methods were: T-statistics [11], information Gain [9], relief [23], gain ratio [9] and chi-squared [37]. Features were evaluated by each classifier indicated in the gene (\(G_{clf}\)): decision tree [32], J48 [29], random tree [4], random forest [4], multilayer perceptron [12], support vector machine (SVM) [33], K-nearest neighbors (KNN) [14] and KStar (K*) [8]. These techniques were applied on each training set and tested using k-folds cross-validation, with \(k=10\).
The structure of the model requires some parameters as inputs to define the best association, such as: population size (P), maximum number of generations or iterations (Iter), selection threshold (t) (representing who will be selected for reproduction—crossover), genetic mutation probability (m) and maximum number of features (MaxF) defined from the initial set of features. The MaxF parameter allows to limit the number of features that constitutes an individual \(G_{id}\). Considering the input parameters, the proposed method processes the information based on population evaluation, selection of the most fit individuals, reproduction (crossover) and mutation.
2.1 Population Evaluation and Selection
Population evaluation consists in calculating the mean accuracy (the fitness function) produced by each individual, based on the selection and classification techniques indicated in their genes. Therefore, for each individual, a selection method \(G_{sel}\) was applied on each of the k training fold and the results were the indexes of the N best features, being N defined by the value drawn for the \(G_{num}\) parameter.
The classifier indicated in \(G_ {clf}\) was trained considering the selected N features. This process was performed for each training file constructed by the \(k = 10\) cross-validation technique. The classification was executed in each correspondent test file, composed by chosen features. Therefore, for each individual \(G_{id}\), L accuracy values were obtained, one for each k training test. The average accuracy \(MeanAcc(G_{id})\) was calculated by applying Eq. 1:
The natural selection behavior proposed by Darwin was considered in the method presented by sorting the accuracy rates (\(MeanAcc(G_{id})\)) and selecting individuals with greater values than the selection parameter t (\(t=0.7\)). This model was developed considering the proposal described by Yang and Honavar [35]. Also, it is important to emphasize that chosen individuals were defined as parents in the next generation, by gathering genes (methods and features) capable of providing an acceptable solution: better combinations of features, selection methods and classifiers.
2.2 Crossover and Mutation
Reproduction is responsible for complementing the population on the current generation with individuals produced from those selected in the previous step. This type of approach simulates the sexual reproduction found in several species in nature. The genetic operation of crossover was implemented using the two-point approach, aiming the search for the best solution by replacing both selection and classification methods. Two-point crossover consists in choosing two locus of a chromosome as points of exchange (or pivots) and alternately making the copy of the genes of the parents for the two children generated. In the chromosome structure used in the model proposed, with the exception of the \(G_{id}\) gene, the other parts were used to determine the next generations. The mutation operator was applied on children to define the next iteration’s population. The mutation operation consisted of a few steps:
-
for each new born individual, a random number \(\alpha \) is drawn to indicate whether the child should be mutated, considering \(\alpha \) \(\in \) \(\mathbb {R}\) \(\mid \) 0 \(\le \) \(\alpha \) \(\le \) 1;
-
if \(\alpha \) > m, being m mutation probability, which was defined as 0.05% [25], the individual is not mutated. Otherwise, \(\alpha \) \(\le \) m, the individual will be mutated.
When an individual is submitted to the mutation process, an index \(\beta \) is drawn to indicate which gene must be mutated. The variable \(\beta \) can assume 1, 2 or 3 indexes, which represent the \(G_{sel}\), \(G_{clf}\) and \(G_{num}\) genes, respectively. Flip mutation was applied on the genes representing lists (\(G_{sel}\) and \(G_{clf}\)), as well as the creep mutation for the gene that indicates a number (\(G_{num}\)). In the flip mutation, a method was replaced by another of the same type. In the creep mutation, a value was subtracted or added to the gene. Considering these mutation processes, the following steps were performed:
-
if \(\beta \) = 1, 2, “take-the-next” strategy was applied on selection methods and classification methods lists.
-
if \(\beta \) = 3, the chosen gene is \(G_{num}\). In this case, a new random number \(\gamma \) is drawn, given by \(\gamma \) = 0 or \(\gamma \) = 1. If \(\gamma \) = 1, \(G_{num}\) is incremented by one. Otherwise (\(\gamma \) = 0), \(G_{num}\) is decremented by one. The maximum value for \(G_{num}\) is delimited by MaxF (maximum number of features).
The procedures previously described was repeated until the maximum number of generations or if 99% (or more) individuals provide a MeanAcc rate equal to 100%. A summary of the proposed method is shown in Algorithm 1.

2.3 Colorectal Database and Feature Set
The tests were performed from features extracted from a dataset of histological colorectal cancer images. They were defined by the method described in [30]. The dataset consists of samples derived from 16 H&E colon histology sections from stage T3 or T4 of colorectal adenocarcinoma. Each section belongs to a patient. Areas with different histological architectures were extracted from the sections and the samples were stained with H&E.
For each input image, features were defined by two Fractal Dimension values \(DF_p\) [18] and \(DF_f\) [27], five lacunarity values (Lac), obtained by area under curve metrics (ARC), skewness (SKW), area ratio (AR), maximum point (MP) and scale of the maximum point SMP, represented by Lac(1) to Lac(5); 14 Haralick texture features (Har) [16], represented by Har(1) up until Har(14), such as angular second moment, correlation and sum of squares; and 15 percolation features (Perc(1) up until Perc(15)) [31], in which ARC, SKW, AR, MP and SMP metrics were also applied for each percolation function, given by cluster average (C), percolating box ratio (Q) and average coverage ratio of the largest cluster (\(\varGamma \)). Mentioned features were also calculated for the curvelet subimages [6]. The curvelets were calculated through observations made on 4 levels of resolution and a sequence of 8 rotation angles. This approach resulted in 41 curvelet subimages for each colorectal image given and a feature set composed by 1.512 features.
3 Results
The feature set was given as input to our method and analyzed from tests defined with different values for P (numbers of individuals) and Iter (iterations or generations). The purpose was to verify the method’s behavior under different situations, as well as identify possible patterns in the context of colorectal images. Results provided by the method are available from Tables 1 and 2, obtained by population values of \(P=50\) and \(P=500\). These values were defined considering works available in the literature [1] [5] and in order to indicate the best combinations in each scenario. The results represent a set of (random) possible solutions involving: selection method, classifier, number of features (NumF) and average accuracy rate (MeanAcc). Area under the ROC curve (AUC) was also collected in each test to complement the performance comparisons of our proposal.
Analyzing the results it is possible to observe considering P = 50, the best result was found with 100 iterations. The solution was indicated by relief (selection method) and random forest (classifier). In this case, the accuracy rate was 87.97%. The best case was determined with a significant number of individuals (P = 500). The highest accuracy rate with the lowest number of features was indicated with 50 iterations. The solution defined by our method provided an accuracy of 90.82%, computed with 29 features, Relief (selection method) and random forest (classifier). In this case, the value of AUC was 0.967. It is important to mention that despite the same accuracy in the tests performed with 100 and 500 iterations, even with indications of the same selection method and classifier, the difference in the number of features is significant.
Colorectal cancer classification from histological images is the subject of several papers available in the literature, such as those described in [20] and [19]. Therefore, a performance overview obtained with our proposal is presented in Table 3, based on AUC rate (which was measured by all the works used for this verification), total features and classification methods.
It is important to observe that direct comparisons cannot be performed to indicate the best approach, since different methodologies and databases were used. Nevertheless, considering the rate provided by our proposal and what was found in the literature, we believe the method is promising and capable of providing an acceptable solution (indication of the highest distinction rate considering the least number of features as possible). Our solution indicated a 0.967 AUC rate, with 29 of 1.512 features, values compatible with important works in the literature directed to the development of CAD systems and colorectal cancer.
One of the advantages of our proposal is identifying the most relevant features and its values (Table 4). It is possible to observe that most features were selected by percolation descriptors and subimages association, totalizing 16 features. Lacunarity attribute was the second most selected type, totalizing 9 features, with measurements obtained (total of 8), mainly of curvelet subimages. Lastly, Haralick’s measures contributed with four metrics. On the other hand, multiscale and multidimensional fractal dimension measurements (\(DF_p\) and \(DF_f\)) were not selected by our strategy to classify colorectal cancer from the H&E images. We believe that this information is important for the CAD system development area. It is possible to observe that area under the curve (ARC), skewness (SKW), area ratio (AR), maximum point (MP) and maximum point scale (SMP) were the most used features for lacunarity and percolation attributes. These metrics were obtained mainly from the combination with curvelet subimages.
4 Conclusion
In this work, a method based on GA capable of finding the best combination of features, selection methods and classifier was proposed in order to provide information for the diagnosis of colorectal cancer from H&E images. This methodology was built from a structured model of evaluation, selection, crossover and mutation. The method presented relevant results. The best solution was determined from 500 individuals and 50 iterations, resulting in 29 features, Relief selection algorithm and random forest classifier. The accuracy rate obtained was 90.82% and the AUC rate was 0.967. Performance was compared to important works available in the literature. The results were relevant, especially when considering the use of comparisons under similar conditions and the number of features considered. As an overview was given from studies developed for colorectal cancer classification, the performance was similar to which is available in the literature, with the differential of discriminating and detailing possible patterns of features indicated for separation of benign and malignant groups of colorectal cancer. In future works we intend to explore different values for the parameters required by our model and types of images. At last, we intend to test our model for pattern recognition in H&E images of lymphomas and breast cancer, with or without normalization of the dyes present in the slides.
References
Al-Rajab, M., Lu, J., Xu, Q.: Examining applying high performance genetic data feature selection and classification algorithms for colon cancer diagnosis. Comput. Methods Programs Biomed. 146, 11–24 (2017)
Alteri, R., Kramer, J., Simpson, S.: Colorectal Cancer Facts and Figures 2014–2016, pp. 1–30. American Cancer Society, Atlanta (2014)
Anbarasi, M., Anupriya, E., Iyengar, N.: Enhanced prediction of heart disease with feature subset selection using genetic algorithm. Int. J. Eng. Sci. Technol. 2(10), 5370–5376 (2010)
Breiman, L.: Random forests. Mach. Learn. 45(1), 5–32 (2001)
Bruderer, E., Singh, J.V.: Organizational evolution, learning, and selection: a genetic-algorithm-based model. Acad. Manag. J. 39(5), 1322–1349 (1996)
Candès, E.J., Donoho, D.L.: New tight frames of curvelets and optimal representations of objects with piecewise \({\rm c}^2\) singularities. Commun. Pure Appl. Math. 57(2), 219–266 (2004)
Chan, H.P., Charles, E., Metz, P., Lam, K., Wu, Y., Macmahon, H.: Improvement in radiologists’ detection of clustered microcalcifications on mammograms. Arbor 1001, 48109–0326 (1990)
Cleary, J.G., Trigg, L.E.: K*: an instance-based learner using an entropic distance measure. In: Machine Learning Proceedings, pp. 108–114. Elsevier (1995)
Dai, J., Xu, Q.: Attribute selection based on information gain ratio in fuzzy rough set theory with application to tumor classification. Appl. Soft Comput. 13(1), 211–221 (2013)
Doi, K.: Computer-aided diagnosis in medical imaging: historical review, current status and future potential. Comput. Med. Imaging Graph. 31(4–5), 198–211 (2007)
Eltoukhy, M.M., Faye, I., Samir, B.B.: A statistical based feature extraction method for breast cancer diagnosis in digital mammogram using multiresolution representation. Comput. Biol. Med. 42(1), 123–128 (2012)
Gardner, M.W., Dorling, S.: Artificial neural networks (the multilayer perceptron)-a review of applications in the atmospheric sciences. Atmos. Environ. 32(14–15), 2627–2636 (1998)
Gonçalves, E.C., Freitas, A.A., Plastino, A.: A survey of genetic algorithms for multi-label classification. In: 2018 IEEE Congress on Evolutionary Computation (CEC), pp. 1–8. IEEE (2018)
Gou, J., Ma, H., Ou, W., Zeng, S., Rao, Y., Yang, H.: A generalized mean distance-based k-nearest neighbor classifier. Expert Syst. Appl. 115, 356–372 (2019)
Gurcan, M.N., et al.: Lung nodule detection on thoracic computed tomography images: preliminary evaluation of a computer-aided diagnosis system. Med. Phys. 29(11), 2552–2558 (2002)
Haralick, R.M.: Statistical and structural approaches to texture. Proc. IEEE 67(5), 786–804 (1979)
IARC: Cancer fact sheets: Colorectal cancer. Technical report, International Agency for Research on Cancer, Lyon, France (2012)
Ivanovici, M., Richard, N., Decean, H.: Fractal dimension and lacunarity of psoriatic lesions-a colour approach. Medicine 6(4), 7 (2009)
Jørgensen, A.S., et al.: Using cell nuclei features to detect colon cancer tissue in hematoxylin and eosin stained slides. Cytometry Part A 91(8), 785–793 (2017)
Kather, J.N., et al.: Multi-class texture analysis in colorectal cancer histology. Sci. Rep. 6, 27988 (2016)
Kečo, D., Subasi, A., Kevric, J.: Cloud computing-based parallel genetic algorithm for gene selection in cancer classification. Neural Comput. Appl. 30(5), 1601–1610 (2018)
Khan, A., Qureshi, A.S., Hussain, M., Hamza, M.Y., et al.: A recent survey on the applications of genetic programming in image processing. arXiv preprint arXiv:1901.07387 (2019)
Kira, K., Rendell, L.A.: A practical approach to feature selection. In: Machine Learning Proceedings, pp. 249–256. Elsevier (1992)
Lu, C., Zhu, Z., Gu, X.: An intelligent system for lung cancer diagnosis using a new genetic algorithm based feature selection method. J. Med. Syst. 38(9), 97 (2014)
Mitchell, M.: An Introduction to Genetic Algorithms. MIT Press, Cambridge (1998)
Muni, D.P., Pal, N.R., Das, J.: Genetic programming for simultaneous feature selection and classifier design (2006)
Nikolaidis, N., Nikolaidis, I., Tsouros, C.: A variation of the box-counting algorithm applied to colour images. arXiv preprint arXiv:1107.2336 (2011)
Paul, D., Su, R., Romain, M., Sébastien, V., Pierre, V., Isabelle, G.: Feature selection for outcome prediction in oesophageal cancer using genetic algorithm and random forest classifier. Comput. Med. Imaging Graph. 60, 42–49 (2017)
Quinlan, J.R.: C4. 5: Programs for Machine Learning. Elsevier, Amsterdam (2014)
Ribeiro, M.G., Neves, L.A., do Nascimento, M.Z., Roberto, G.F., Martins, A.S., Tosta, T.A.A.: Classification of colorectal cancer based on the association of multidimensional and multiresolution features. Expert Syst. Appl. 120, 262–278 (2019). https://doi.org/10.1016/j.eswa.2018.11.034, http://www.sciencedirect.com/science/article/pii/S0957417418307541
Roberto, G.F.: Features based on the percolation theory for quantification of non-hodgkin lymphomas. Comput. Biol. Med. 91, 135–147 (2017)
Safavian, S.R., Landgrebe, D.: A survey of decision tree classifier methodology. IEEE Trans. Syst. Man Cybern. 21(3), 660–674 (1991)
Vapnik, V.N.: An overview of statistical learning theory. IEEE Trans. Neural Netw. 10(5), 988–999 (1999)
Whitley, D.: A genetic algorithm tutorial. Stat. Comput. 4(2), 65–85 (1994)
Yang, J., Honavar, V.: Feature subset selection using a genetic algorithm. In: Liu, H., Motoda, H. (eds.) Feature Extraction, Construction and Selection, vol. 453, pp. 117–136. Springer, Heidelberg (1998). https://doi.org/10.1007/978-1-4615-5725-8_8
Yu, S., Guan, L.: A CAD system for the automatic detection of clustered microcalcifications in digitized mammogram films. IEEE Trans. Med. Imaging 19(2), 115–126 (2000)
Zheng, Z., Wu, X., Srihari, R.: Feature selection for text categorization on imbalanced data. ACM SIGKDD Explor. Newslett. 6(1), 80–89 (2004)
Acknowledgments
The authors gratefully acknowledge the financial support of National Council for Scientific and Technological Development CNPq (Grants #427114/2016-0, #304848/2018-2, #430965/2018-4 and #313365/2018-0), the State of Minas Gerais Research Foundation - FAPEMIG (Grant #APQ-00578-18).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Taino, D.F. et al. (2019). A Model Based on Genetic Algorithm for Colorectal Cancer Diagnosis. In: Nyström, I., Hernández Heredia, Y., Milián Núñez, V. (eds) Progress in Pattern Recognition, Image Analysis, Computer Vision, and Applications. CIARP 2019. Lecture Notes in Computer Science(), vol 11896. Springer, Cham. https://doi.org/10.1007/978-3-030-33904-3_47
Download citation
DOI: https://doi.org/10.1007/978-3-030-33904-3_47
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-33903-6
Online ISBN: 978-3-030-33904-3
eBook Packages: Computer ScienceComputer Science (R0)
