
Abstract
Gene coexpression patterns carry rich information of complex brain structures and functions. Characterization of these patterns in an unbiased and integrated manner will illuminate the higher order transcriptome organization and offer molecular foundations of functional circuitry. Here we demonstrate a data-driven method that can effectively extract coexpression networks from transcriptome profiles using the Allen Mouse Brain Atlas dataset. For each of the obtained networks, both genetic compositions and spatial distributions in brain volume are learned. A simultaneous knowledge of precise spatial distributions of specific gene as well as the networks the gene plays in and the weights it carries can bring insights into the molecular mechanism of brain formation and functions. Gene ontologies and the comparisons with published data reveal interesting functions of the identified coexpression networks, including major cell types, biological functions, brain regions, and/or brain diseases.
Y. Li and H. Chen—Co-first Authors.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Gene coexpression patterns carry rich amount of valuable information regarding enormously complex cellular processes. Previous studies have shown that genes displaying similar expression profiles are very likely to participate in the same biological processes [1]. The gene coexpression network (GCN), offering an integrated and effective representation of gene interactions, has shown advantages in deciphering the biological and genetic mechanisms across species and during evolution [2]. In addition to revealing the intrinsic transcriptome organizations, GCNs have also demonstrated superior performance when they are used to generate novel hypotheses for molecular mechanisms of diseases because many disease phenotypes are a result of dysfunction of complex network of molecular interactions [3].
Various proposals have been made to identify the GCNs, including the classical clustering methods [4, 5] and those applying network concepts and models to describe gene-gene interactions [6]. Given the high dimensionality of genetic data and the urgent need of making comparisons to unveil the changes or the consensus between subjects, one common theme of these methods is dimension reduction. Instead of analyzing the interactions between over ten thousands of genes, the groupings of data by its co-expression patterns can considerably reduce the complexity of comparisons from tens of thousands of genes to dozens of networks or clusters while preserving the original interactions.
Along the line of data-reduction, we proposed dictionary learning and sparse coding (DLSC) algorithm for GCN construction. DLSC is an unbiased data-driven method that learns a set of new dictionaries from the signal matrix so that the original signals can be represented in a sparse and linear manner. Because of the sparsity constraint, the dimensions of genetic data can be significantly reduced. The grouping by co-expression patterns are encoded in the sparse coefficient matrix with the assumption that if two genes use same dictionary to represent their original signals, their gene expressions must share similar patterns, and thereby considered ‘coexpressed’. The proposed method overcomes the potential issues of overlooking multiple roles of regulatory domains in different networks that are seen in many clustering methods [3] because DLSC does not impose the bases be orthogonal so that one gene can be claimed by multiple networks. More importantly, for each of the obtained GCNs, both genetic compositions and spatial distributions are learned. A simultaneous knowledge of precise distributions of specific gene as well as the networks the gene plays in and the weights it carries can bring insights into the molecular mechanism of brain formation and functions.
In this study the proposed framework was applied on Allen Mouse Brain Atlas (AMBA) [7], which surveyed over 20,000 genes expression patterns in C57BL6 J mouse brain using in situ hybridization (ISH). One major advantage of ISH is the ability of preserving the precise spatial distribution of genes. This powerful dataset, featured by the whole-genome scale, cellular resolution and anatomical coverage, has made it possible for a holistic understanding of the molecular underpinnings and related functional circuitry. Using AMBA, the GCNs identified by DLSC showed significant enrichment for major cell types, biological functions, anatomical regions, and/or brain disorders, which holds promises to serve as foundations to explore different cell types and functional processes in diseased and healthy brains.
2 Methods
2.1 Experimental Setup
We downloaded the 4,345 3D volumes of expression energy of coronal sections and the Allen Reference Atlas (ARA) from the website of AMBA (http://mouse.brain-map.org/). The ISH data were collected in tissue sections, then digitally processed, stacked, registered, gridded, and quantified - creating 3D maps of gene “expression energy” at 200 micron resolution. Coronal sections are chosen because they registered more accurately to the reference model than the counterparts of sagittal sections. Each 3D volume is composed by 67 slices with a dimension of 41 × 58.
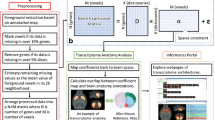
As the ISH data is acquired by coronal slice before they were stitched and aligned into a complete 3D volume, in spite of extensive preprocessing steps, quite significant changes in average expression levels of the same gene in the adjacent slices are observed. To alleviate the artifacts due to slice handling and preprocessing, we decided to study the coexpression networks slice by slice. The input of the pipeline are the expression grids of one of 67 coronal slices (Fig. 1a). A preprocessing module (Fig. 1b) is first applied to handle the foreground voxels with missing data (−1 in expression energy). Specifically, this module includes extraction, filtering and estimation steps. First, the foreground voxels of the slice based on ARA were extracted. Then the genes of low variance or with missing values in over 20 % of foreground voxels were excluded. A similar filtering step is also applied to remove voxels in which over 20 % genes do not have data. Most missing values were resolved in the filtering steps. The remaining missing values were recursively estimated as the mean of foreground voxels in its 8 neighborhood until all missing values were filled. After preprocessing, the cleaned expression energies were organized into a matrix and sent to DLSC (Fig. 1c). In DLSC, the gene expression matrix is factorized into a dictionary matrix D and a coefficient matrix α. These two matrices encode the distribution and composition of GCN (Fig. 1d–e) and will be further analyzed.

Computational pipeline for constructing GCNs. (a) Input is one slice of 3D expression grids of all genes. (b) Raw ISH data preprocessing step that removes unreliable genes and voxels and estimates the remaining missing data. (c) Dictionary learning and sparse coding of ISH matrix with sparse and non-negative constraints on coefficient α matrix. (d) Visualization of spatial distributions of GCNs. (e) Enrichment analysis of GCNs.
2.2 Dictionary Learning and Sparse Coding
DLSC is an effective method to achieve a compressed and succinct representation for ideally all signal vectors. Given a set of M-dimensional input signals X = [x1,…,xN] in \( {\mathbb{R}}^{M \times N} \), learning a fixed number of dictionaries for sparse representation of X can be accomplished by solving the following optimization problem. As discussed later that each entry of α indicates the degree of conformity of a gene to a coexpression pattern, a non-negative constraint was added to the \( \ell_{1} \) regularization.
where \( \varvec{D} \in {\mathbb{R}}^{N \times K} \) is the dictionary matrix, \( \varvec{\alpha}\in {\mathbb{R}}^{K \times M} \) is the corresponding coefficient matrix, λ is a sparsity constraint factor and indicates each signal has fewer than λ items in its decomposition, \( \left\| \varvec{*} \right\|_{1} ,\left\| \varvec{*} \right\|_{2} \) are the summation of \( \ell_{1} {\text{and }}\ell_{2} \) norm of each column. \( \left\| {\varvec{X} - \varvec{D} \times\varvec{\alpha}} \right\|_{{^{2} }}^{2} \) denotes the reconstruction error.
In practice, gene expression grids are arranged into a matrix \( \varvec{X} \in {\mathbb{R}}^{M \times N} \), such that rows correspond to foreground voxels and columns correspond to genes (Fig. 1c). After normalizing each column by its Frobenius norm, the publicly available online DLSC package [8] was applied to solve the matrix factorization problem proposed in Eq. (1). Eventually, X is represented as sparse combinations of learned dictionary atoms D. Each column in D is one dictionary consisted of a set of voxels. Each row in α details the coefficient of each gene in a particular dictionary. The key assumptions of enforcing the sparseness is that each gene is involved in a very limited number of gene networks. The non-negativity constraint on α matrix imposes that no genes with the opposite expression patterns placed in the same network. In the context of GCN construction, we consider that if two genes use the same dictionary to represent the original signals, then the two genes are ‘coexpressed’ in this dictionary. This set-up has several benefits. First, both the dictionaries and coefficients are learnt from the data and therefore intrinsic to the data. Second, the level of coexpressions are quantifiable and not only comparable within one dictionary, but the entire α matrix.
Further, if we consider each dictionary as one network, the corresponding row of α matrix contains all genes that use this dictionary for sparse representation, or ‘coexpressed’. Each entry of α measures the extent to which this gene conforms to the coexpression pattern described by the dictionary atom. Therefore, this network, denoted as the coexpression network, is formed. Since the dictionary atom is composed of multiple voxels, by mapping each atom in D back to the ARA space, we can visualize the spatial patterns of the coexpressed networks. Combining information from both D and α matrices, we would obtain a set of intrinsically learned GCNs with the knowledge of both their anatomical patterns and gene compositions. As the dictionary is the equivalent of network, these two terms will be used interchangeably.
The choice of number of dictionaries and the regularization parameter λ are crucial to an effective sparse representation. The final goal here is a set of parameters that result in a sparse and accurate representation of the original signal while achieving the highest overlap with the ground truth - the known anatomy. A grid search of parameters is performed using three criteria: (1) reconstruction error; (2) mutual information between the dictionaries and ARA; (3) the density of α matrix measured by the percentage of none-zero-valued elements in α. As different number of genes are expressed in different slices, instead of a fixed number of dictionaries, a gene-dictionary ratio, defined as the ratio between the number of genes expressed and the number of dictionaries, is used. Guided by these criteria, λ = 0.5 and gene-dictionary ratio of 100 are selected as the optimal parameters.
2.3 Enrichment Analysis of GCNs
GCNs were characterized based on common gene ontology (GO) categories (molecular function, biological process, cellular component), using Database for Annotation, Visualization and Integrated Discovery (DAVID) [9]. Enrichment analysis was performed by cross-referencing with published lists of genes related to cell type markers, known and predicted lists of disease genes, specific biological functions etc. This list consists of 32 publications and is downloaded from [10]. Significance was assessed using one-sided Fisher’s exact test with a threshold of p < 0.01.
3 Results
DLSC allows readily interpretable results by plotting the spatial distributions of GCNs. A visual inspection showed a set of spatially contiguous clusters partitioning the slice (Fig. 2a,e). Many formed clusters correspond to one or more canonical anatomical regions, providing an intuitive validation to the approach.

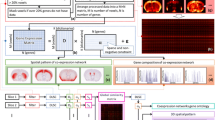
Visualization of spatial distribution of GCNs and the corresponding raw ISH data. On the left are the slice ID and GCN ID. The second columns are the spatial maps of two GCNs, one for each slice, followed by the ISH raw data of 3 representative genes. Gene acronyms and the weights in the GCN are listed at the bottom. The weights indicate the extent to which a gene conforms to the GCN.
We will demonstrate the effectiveness of the DLSC by showing that the GCNs are mathematically valid and biologically meaningful. Since the grouping of genes is purely based on their expression patterns, a method with good mathematical ability will make the partitions so that the expression patterns of the genes are similar within group and dissimilar between groups. One caveat is that one gene may be expressed in multiple cell types or participate in multiple functional pathways. Therefore, maintaining the dissimilarity between groups may not be necessary. At the same time, the method should also balance the biological ability of finding functionally enriched networks. To show as an example, slice 27 and 38 are analyzed and discussed in depth due to its good anatomical coverage of various brain regions. Using a fixed gene-dictionary ratio of 100, 29 GCNs were identified for slice 27 and 31 GCNs were constructed on slice 38.
3.1 Validation Against Raw ISH Data
One reliable way to examine whether the expression patterns are consistent within a GCN is to visually inspect the raw ISH data where the GCNs are derived. As seen in Fig. 2, in general the ISH raw data match well with the respective spatial map. In GCN 22 of slice 27, the expression peaks at hypothalamus and extends to the substantia innominate and bed nuclei of the stria teminais (Fig. 2a). All three genes showed strong signals in these areas in the raw data (Fig. 2b–d). Similarly, the expressions patterns for GCN 6 in slice 38 centered at the field CA1 (Fig. 2e) and all three genes showed significantly enhanced signals in the CA1 region compared with other regions (Fig. 2f–h). Relatedly, the weight in the parentheses is a measure of the degree to which a gene conforms to the coexpression patterns. With a decreasing weight, the resemblance of the raw data to the spatial map becomes weaker. One example is the comparison between Zkscanl6 and the other two genes. Weaker signals were found in lateral septal nucleus (Fig. 2b–d red arrow), preoptic area (Fig. 2b–d blue arrow), and lateral olfactory tract (Fig. 2b–d green arrows) in Zkscanl6. On the other hand, the spatial map of GCN 6 of slice 38 features an abrupt change of expression levels at the boundaries of field CA1 and field CA2. This feature is seen clearly in Fibcd (Fig. 2 f red arrows) and Arhgap12 (Fig. 2g red arrows), but not Osbp16 (Fig. 2h red arrows). Also, the spatial map shows an absence of expression in the dental gyrus (DG) and field CA3. However, Arhgap12 displays strong signals at DG (Fig. 2g blue arrows) and Osbp16 shows high expressions in both DG and CA3 (Fig. 2h green arrows). The decreased similarity agrees well with the declining weights. Overall, we have demonstrated a good agreement between the ISH raw data and the corresponding spatial map. The level of agreement is correctly measured by the weight. These results visually validate the mathematical ability of DLSC in grouping genes with similar expression patterns.
3.2 Enrichment Analysis of GCNs
Enrichment analysis using GO terms and existing published gene lists [10] provided exciting biological insights for the constructed GCNs. We roughly categorize the networks into four types for the convenience of presentation. In fact, one GCN often falls into multiple categories as these categories characterize GCNs from different perspectives. A comparison with the gene lists generated using purified cellular population [11, 12] indicates that GCN1 (Fig. 3a), GCN5 (Fig. 3b), GCN28 (Fig. 3c), GCN25 (Fig. 3d) of slice 27 are significantly enriched with markers of oligodendrocytes, astrocytes, neurons and interneurons, with the p-values to be 1.1 × 10−7, 1.7 × 10−8, 2.5 × 10−3, 15 × 10−10 respectively. The findings have been not only confirmed by several other studies using microarray and ISH data, but also corroborated by the GO terms. For example, two significant GO term in GCN1 is myelination (p = 5.7 × 10−4) and axon ensheathment (p = 2.5 × 10−5), which are featured functions for oligodendrocyte, with established markers such as Mbp, Serinc5, Ugt8a. A visualization of the spatial map also offers a useful complementary source. For example, the fact that GCN5 (Fig. 3b) locates at the lateral ventricle, where the subventricular zone is rich with astrocytes, confirms its enrichment in astrocyte.

Visualization of spatial distribution of GCNs enriched for major cell types, particular brain regions and function/disease related genes. In each panel, top row: Slice ID and GCN ID; second row: spatial map; third row: sub-category; fourth row: highly weighted genes in the sub-category.
In addition to cell type specific GCNs, we also found some GCNs remarkably selective for particular brain regions, such as GCN3 (Fig. 3e) in CA1, GCN5 (Fig. 3f) in thalamus, GCN11 (Fig. 3g) in hypothalamus and GCN16 (Fig. 3h) in caudeputaman. Other GCNs with more complex anatomical patterning revealed close associations to biological functions and brain diseases. The GCNs associated with ubiquitous functions such as ribosomal (Fig. 3j) and mitochondrial functions (Fig. 3k) have a wide coverage of brain. A functional annotation suggested GCN12 of slice 27 is highly enriched for ribosome pathway (p = 6.3 × 10−5). As to GCN21 on the same slice, besides mitochondrial function (p = 1.5 × 10−8), it also enriches in categories including neuron (p = 5.4 × 10−8) and postsynaptic proteins (p = 6.3 × 10−8) comparing with literatures [10]. One significant GO term synaptic transmission (p = 1.1 × 10−5) might add possible explanations to the strong signals in the cortex regions. GCN13 of slice 38 (Fig. 3i) showed strong associations with genes that found downregulated in Alzheimer’s disease. Comparisons with Autism susceptible genes generated from microarray and high-throughput RNA-sequencing data [13] indicates GCN 24 of slice 27’s association (p = 1.0 × 10−3) (Fig. 3h). Despite slightly lower weights, the most significant three genes Met, Pip5k1b, Avpr1a, have all been reported altered in Autism patients [13].
4 Discussion
We have presented a data-driven framework that can derive biologically meaningful GCNs from the gene expression data. Using the rich and spatially-resolved ISH AMBA data, we found a set of GCNs that are significantly enriched for major cell types, anatomical regions, biological pathways and/or brain diseases. The highlighted advantage of this method is its capability of visualizing the spatial distribution of the GCNs while knowing the gene constituents and the weights they carry in the network. Although the edges in the network are not explicitly stated, it does not impact the interpretations of the GCNs biologically.
In future work, new strategies will be developed to integrate the gene-gene interactions on a single slice and to construct brain-scale GCNs. These GCNs can offer new insights in multiple applications. For example, GCNs can serve as a baseline network to enable comparisons across different species to understand brain evolution. Also, charactering GCNs of brains at different stages can generate new hypotheses of brain formation process. When the GCNs are correlated with neuroimaging measurements as brain phenotypes, we are able to make new associations between the molecular scales and the macro scale measurement and advance the understanding of how genetic functions regulate and support brains structures and functions, as well as finding new genetic variants that might account for the variations in brain structure and functions.
References
Tavazoie, S., Hughes, J.D., et al.: Systematic determination of genetic network architecture. Nat. Genet. 22, 281–285 (1999)
Stuart, J.M.: A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003)
Gaiteri, C., Ding, Y., et al.: Beyond modules and hubs: the potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes. Brain Behav. 13, 13–24 (2014)
Bohland, J.W., Bokil, H., et al.: Clustering of spatial gene expression patterns in the mouse brain and comparison with classical neuroanatomy. Methods 50, 105–112 (2010)
Eisen, M.B., Spellman, P.T., et al.: Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 95, 12930–12933 (1999)
Langfelder, P., Horvath, S.: WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008)
Lein, E.S., Hawrylycz, M.J., Ao, N., et al.: Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007)
Mairal, J., Bach, F., et al.: Online learning for matrix factorization and sparse coding. J. Mach. Learn. Res. 11, 19–60 (2010)
Dennis, G., Sherman, B.T., et al.: DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3 (2003)
Miller, J.A., Cai, C., et al.: Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinform. 12, 322 (2011)
Cahoy, J., Emery, B., Kaushal, A., et al.: A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neuronsci. 28, 264–278 (2004)
Winden, K.D., Oldham, M.C., et al.: The organization of the transcriptional network in specific neuronal classes. Mol. Syst. Biol. 5, 1–18 (2009)
Voineagu, I., Wang, X., et al.: Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474(7351), 380–384 (2011)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this paper
Cite this paper
Li, Y. et al. (2016). Discover Mouse Gene Coexpression Landscape Using Dictionary Learning and Sparse Coding. In: Ourselin, S., Joskowicz, L., Sabuncu, M., Unal, G., Wells, W. (eds) Medical Image Computing and Computer-Assisted Intervention – MICCAI 2016. MICCAI 2016. Lecture Notes in Computer Science(), vol 9900. Springer, Cham. https://doi.org/10.1007/978-3-319-46720-7_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-46720-7_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46719-1
Online ISBN: 978-3-319-46720-7
eBook Packages: Computer ScienceComputer Science (R0)