
Abstract
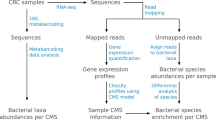
The huge amount of genomic and transcriptomic data obtained to characterize human diversity can also be exploited to indirectly gather information on the human microbiome. Here we present the pipeline QmihR designed to identify and quantify the abundance of known microbiome communities and to search for new/rare pathogenic species in RNA-seq datasets. We applied QmihR to 36 RNA-seq tumor tissue samples from Ukrainian gastric carcinoma patients available in TCGA, in order to characterize their microbiome and check for efficiency of the pipeline. The microbes present in the samples were in accordance to published data in other European datasets, and the independent BLAST evaluation of microbiome-aligned reads confirmed that the assigned species presented the highest BLAST match-hits. QmihR is available at GitHub (https://github.com/Pereira-lab/QmihR).
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others

References
Cho, I., Blaser, M.J.: The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13, 260–270 (2012)
Turnbaugh, P.J., Ley, R.E., Hamady, M., Fraser-Liggett, C., Knight, R., Gordon, J.I.: The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature 449, 804–810 (2007)
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K.S., Manichanh, C., Nielsen, T., Pons, N., Levenez, F., Yamada, T., Mende, D.R., Li, J., Xu, J., Li, S., Li, D., Cao, J., Wang, B., Liang, H., Zheng, H., Xie, Y., Tap, J., Lepage, P., Bertalan, M., Batto, J.-M., Hansen, T., Le Paslier, D., Linneberg, A., Nielsen, H.B., Pelletier, E., Renault, P., Sicheritz-Ponten, T., Turner, K., Zhu, H., Yu, C., Li, S., Jian, M., Zhou, Y., Li, Y., Zhang, X., Li, S., Qin, N., Yang, H., Wang, J., Brunak, S., Dore, J., Guarner, F., Kristiansen, K., Pedersen, O., Parkhill, J., Weissenbach, J., Bork, P., Ehrlich, S.D., Wang, J.: A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010)
Human Microbiome Project Consortium: Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012)
Bäckhed, F., Ley, R.E., Sonnenburg, J.L., Peterson, D.A., Gordon, J.I.: Host-bacterial mutualism in the human intestine. Science 307, 1915–1920 (2005)
Thomas, R.M., Jobin, C.: The microbiome and cancer: is the ‘Oncobiome’ mirage real? Trends Cancer 1, 24–35 (2015)
Brawner, K.M., Morrow, C.D., Smith, P.D.: Gastric microbiome and gastric cancer. Cancer J. 20, 211–216 (2014). (Sudbury, Mass)
Zhernakova, A., Kurilshikov, A., Bonder, M.J., Tigchelaar, E.F., Schirmer, M., Vatanen, T., Mujagic, Z., Vila, A.V., Falony, G., Vieira-Silva, S., Wang, J., Imhann, F., Brandsma, E., Jankipersadsing, S.A., Joossens, M., Cenit, M.C., Deelen, P., Swertz, M.A., Weersma, R.K., Feskens, E.J., Netea, M.G., Gevers, D., Jonkers, D., Franke, L., Aulchenko, Y.S., Huttenhower, C., Raes, J., Hofker, M.H., Xavier, R.J., Wijmenga, C., Fu, J.: Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016)
Weinstein, J.N., Collisson, E.A., Mills, G.B., Shaw, K.R.M., Ozenberger, B.A., Ellrott, K., Shmulevich, I., Sander, C., Stuart, J.M., Cancer Genome Atlas Research Network.: The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45(10), 1113–1120 (2013)
Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., Hasz, R., Walters, G., Garcia, F., Young, N., Foster, B.: The genotype-tissue expression (GTEx) project. Nat. Genet. 45(6), 580–585 (2013)
Samuels, D.C., Han, L., Li, J., Quanghu, S., Clark, T.A., Shyr, Y., Guo, Y.: Finding the lost treasures in exome sequencing data. Trends Genet. 29, 593–599 (2013)
Chandramohan, R., Wu, P.Y., Phan, J.H., Wang, M.D.: Benchmarking RNA-seq quantification tools. In: 35th Annual International Conference of the IEEE, Engineering in Medicine and Biology Society (EMBC), pp. 647–650 (2013)
Dicksved, J., Lindberg, M., Rosenquist, M., Enroth, H., Jansson, J.K., Engstrand, L.: Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J. Med. Microbiol. 58(4), 509–516 (2009)
Bolger, A.M., Lohse, M., Usadel, B.: Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014)
Williams, C.R., Baccarella, A., Parrish, J.Z., Kim, C.C.: Trimming of sequence reads alters RNA-seq gene expression estimates. BMC Bioinform. 17, 103 (2016)
Langmead, B., Salzberg, S.L.: Fast gapped-read alignment with Bowtie 2. Nat Meth 9, 357–359 (2012)
Li, B., Dewey, C.N.: RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 12, 323 (2011)
Bray, N.L., Pimentel, H., Melsted, P., Pachter, L.: Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34(5), 525–527 (2016)
Tang, K.W., Alaei-Mahabadi, B., Samuelsson, T., Lindh, M., Larsson, E.: The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 4, 2513 (2013)
Acknowledgements
We wish to thank TCGA for the access provided to the protected data used in this work. Funds were guaranteed by the project “Advancing cancer research: from basic knowledge to application”; NORTE-01-0145-FEDER-000029; “Projetos Estruturados de I&D&I”, funded by Norte 2020 – Programa Operacional Regional do Norte. I3S is financed by FEDER - Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020 - Competitiveness and Internationalization Operational Programme (POCI), Portugal 2020, and by Portuguese funds through FCT/Ministério da Ciência, Tecnologia e Inovação in the framework of the project “Institute for Research and Innovation in Health Sciences” (POCI-01-0145-FEDER-007274).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Cavadas, B., Ferreira, J., Camacho, R., Fonseca, N.A., Pereira, L. (2017). QmihR: Pipeline for Quantification of Microbiome in Human RNA-seq. In: Fdez-Riverola, F., Mohamad, M., Rocha, M., De Paz, J., Pinto, T. (eds) 11th International Conference on Practical Applications of Computational Biology & Bioinformatics. PACBB 2017. Advances in Intelligent Systems and Computing, vol 616. Springer, Cham. https://doi.org/10.1007/978-3-319-60816-7_21
Download citation
DOI: https://doi.org/10.1007/978-3-319-60816-7_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60815-0
Online ISBN: 978-3-319-60816-7
eBook Packages: EngineeringEngineering (R0)