
Abstract
Large-scale data processing techniques, currently known as Big-Data, are used to manage the huge amount of data that are generated by sequencers. Although these techniques have significant advantages, few biological applications have adopted them. In the Bioinformatic scientific area, Multiple Sequence Alignment (MSA) tools are widely applied for evolution and phylogenetic analysis, homology and domain structure prediction. Highly-rated MSA tools, such as MAFFT, ProbCons and T-Coffee (TC), use the probabilistic consistency as a prior step to the progressive alignment stage in order to improve the final accuracy. In this paper, a novel approach named PPCAS (Probabilistic Pairwise model for Consistency-based multiple alignment in Apache Spark) is presented. PPCAS is based on the MapReduce processing paradigm in order to enable large datasets to be processed with the aim of improving the performance and scalability of the original algorithm.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


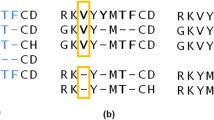
Notes
- 1.
Given a MSA containing three sequences x, y, and z, if position \(x_i\) aligns with position \(z_k\) and position \(z_k\) aligns with \(y_j\) in the projected x-z and z-y alignments, then to be consistent the \(x_i\) must align with \(y_j\) in the projected x-y alignment.
- 2.
PPCAS is available on https://github.com/jllados/PPCAS.
References
Abramova, V., Bernardino, J., Furtado, P.: Which NoSQL database? A performance overview. Open J. Databases (OJDB) 1(2), 17–24 (2014)
Dean, J., Ghemawat, S.: MapReduce: a flexible data processing tool. Commun. ACM 53(1), 72–77 (2010)
Di Tommaso, P., Moretti, S., Xenarios, I., Orobitg, M., Montanyola, A., Chang, J.-M., Taly, J.-F., Notredame, C.: T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39(2), 13–17 (2011)
Do, C.B., Mahabhashyam, M.S., Brudno, M., Batzoglou, S.: ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 15(2), 330–340 (2005)
Gotoh, O.: Heuristic Alignment Methods. Multiple Sequence Alignment Methods, vol. 1079, pp. 29–43. Springer, Heidelberg (2014)
Katoh, K., Standley, D.M.: MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30(4), 772–780 (2013)
Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J., Higgins, D.G.: Clustal W and Clustal X version 2.0. Bioinformatics 23(21), 2947–2948 (2007)
Li, H., Durbin, R.: Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14), 1754–1760 (2009)
Mount, D.W.: Comparison of the PAM and BLOSUM amino acid substitution matrices. Cold Spring Harbor Protoc. 6 (2008). doi:10.1101/pdb.ip59
Miyazawa, S.: A reliable sequence alignment method based on probabilities of residue correspondences. Protein Eng. Des. Sel. 8(10), 999–1009 (1995)
Myers, E.W., Miller, W.: Optimal alignments in linear space. Bioinformatics 4(1), 11–17 (1988)
Nguyen, K., Guo, X., Pan, Y.: Multiple sequences alignment algorithms. In: Multiple Biological Sequence Alignment Scoring Functions, Algorithms and Applications (2016)
Nguyen, T., Shi, W., Ruden, D.: CloudAligner: a fast and full-featured MapReduce based tool for sequence mapping. BMC Res. Notes 4(1), 171 (2011)
Notredame, C., Holm, L., Higgins, D.G.: COFFEE: an objective function for multiple sequence alignments. Bioinformatics 14(5), 407–422 (1998)
Pireddu, L., Leo, S., Zanetti, G.: SEAL: a distributed short read mapping and duplicate removal tool. Bioinformatics 27(15), 2159–2160 (2011)
Sadasivam, G., Baktavatchalam, G.: A novel approach to Multiple Sequence Alignment using hadoop data grids. Int. J. Bioinform. Res. Appl. 6(5), 472–483 (2010)
Sakr, S.: Big data processing stacks. IT Prof. 19(1), 34–41 (2017)
Schatz, M.: CloudBurst: highly sensitive read mapping with MapReduce. Bioinformatics 25(11), 1363–1369 (2009)
Sievers, F., Dineen, D., Wilm, A., Higgins, D.G.: Making automated multiple alignments of very large numbers of protein sequences. Bioinformatics 29(8), 989–995 (2013)
Smith, A.D., Xuan, Z., Zhang, M.Q.: Using quality scores and longer reads improves accuracy of Solexa read mapping. BMC Bioinform. 9(1), 128 (2008)
Subramanian, A.R., Weyer-Menkhoff, J., Kaufmann, M., Morgenstern, B.: DIALIGN-T: an improved algorithm for segment-based multiple sequence alignment. BMC Bioinform. 6(1), 66 (2005)
Thompson, J.D., Koehl, P., Ripp, R., Poch, O.: BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins Struct. Funct. Bioinf. 61(1), 127–136 (2005)
Zhang, Y., Cao, T., Li, S., Tian, X., Yuan, L., Jia, H., Vasilakos, A.V.: Parallel processing systems for big data: a survey. Proc. IEEE 104(11), 2114–2136 (2016)
Zou, Q.: Survey of MapReduce frame operation in bioinformatics. Brief. Bioinform. 15(4), 637–647 (2014)
Zou, Q., Hu, Q., Guo, M., Wang, G.: HAlign: fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 31(15), 2475–2481 (2015)
Acknowledgments
This work was supported by the MEyC-Spain [contract TIN2014-53234-C2-2-R].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Lladós, J., Guirado, F., Cores, F. (2017). PPCAS: Implementation of a Probabilistic Pairwise Model for Consistency-Based Multiple Alignment in Apache Spark. In: Ibrahim, S., Choo, KK., Yan, Z., Pedrycz, W. (eds) Algorithms and Architectures for Parallel Processing. ICA3PP 2017. Lecture Notes in Computer Science(), vol 10393. Springer, Cham. https://doi.org/10.1007/978-3-319-65482-9_45
Download citation
DOI: https://doi.org/10.1007/978-3-319-65482-9_45
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-65481-2
Online ISBN: 978-3-319-65482-9
eBook Packages: Computer ScienceComputer Science (R0)