
Abstract
The fundamental of complete interaction system of all living cell is protein- protein interactions (PPI). A protein-protein interactions network (PPIN) can be viewed as an intricate system of proteins. The proteins are linked by interactions between themselves. In this work, we developed a new algorithm to find largest quasi-cliques in human PPIN. We also identify significant clusters of proteins for subsequent pathway analysis. In the current experimental setup, we have mined 49 quasi-cliques from the human PPIN, with the largest quasi-clique having size 29. Each of these protein clusters are analysed with KEGG pathway analysis. The algorithm has been compared with the state-of-the art available in this field. We observe that our method is better than other methods available in this domain and finds larger quasi-cliques with higher size.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Proteins interact with other proteins to accomplish biological functions. The transient or more permanent complexes are formed due to such interactions. These interaction networks facilitate biological processes. To study its biological function, it is essential to recognize its probable interaction with other proteins. A PPI network can be thought as a complex system of proteins base on interactions between themselves. Wagner et al. [1] have demonstrated the PPI network as an undirected graph. The nodes are used to represent proteins and edges are used to represent the interaction among proteins. A substantial biological knowledge at the molecular level of interacting proteins can be obtained by PPINs [2]. Important directions for study of biological pathways and protein function can be obtained by mining these networks [3].
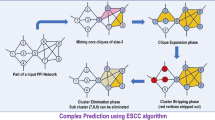
In order to discover the cluster of protein complexes in PPINs, lots of techniques have been used by researchers. These include clustering of sub-graph, finding dense regions [4,5,6], or clique finding [7]. The concept of maximum quasi-clique problem (MQP) is introduced by Matsuda et al. [8]. It is a constrained association of vertices in a graph. It leads to a \( \upgamma \)-quasi-clique.
Subsequently, Pie et al. [9] have proposed a mining algorithm (\( {\text{Crochet}} \)) to discover all quasi-cliques. Further, they have upgraded their algorithm and proposed \( Crochet^{ + } \) for the same purpose [10]. These methods have some restrictions for discovering all quasi-cliques. Brunato et al. [11] have proposed another definition for quasi-clique with a pair of parameters. Bhattacharyya et al. [12] have presented an algorithm to find the biggest quasi-cliques in PPIN of Homo sapiens.
In view of above facts, it is evident that there is an increasing necessity to localize those significant clusters of protein in an interaction network. The existing algorithms for determination of quasi-cliques are not sufficient to address the complexity of many networks. In case of homo-sapiens, simple analysis of tightly coupled cliques from the protein-protein networks may not be sufficient for investigation of key disease pathways. Therefore, we have tried to relax the constraints in an attempt to find all possible maximal quasi-cliques in the networks. The work presented here is found to be computationally efficient from the earlier works Quick [13] and Cocain [14] and the experimental results validates our claims.
In this work, we attempt to search for the largest PPI cluster using a new quasi-clique algorithm (qCliP). In the following, we first described the important preliminaries. After that method is presented along with detailed pathway analysis.
2 Methods
Here, some basic definitions and preliminaries of Maximal Quasi-clique Problem is first described. The graph indicate undirected labelled simple graph. A graph \( G \) is defined by tuples \( \left( {V,E} \right) \), where \( V \) is set of vertices and E is set of edges in between the pair of vertices. Our objective is to search for all possible maximal quasi-cliques in PPINs. A PPIN can be defined by tuples \( \left( {P,I } \right) \), where \( {\text{P}} \) denotes protein set and \( I \) denotes set of interactions. So, we have direct analogy between \( \left( {V, E} \right) \) and \( \left( {P,I} \right) \).
\( \gamma \) -quasi-clique graph: For \( \left( {0 < \gamma \le 1} \right) \), if each vertices of the graph \( G \) has at least degree \( \left\lceil {\gamma \times \left( {\left| V \right| - 1} \right)} \right\rceil \) then such graph \( G \) is called \( \gamma \)-quasi-clique graph.
An algorithm to find the largest quasi-clique is presented in the following. The proposed method finds quasi-cliques in large protein-protein interaction networks.

3 Results and Discussion
In this work, we have first started with 1831 distinct human proteins involving 2252 interactions (dip20100614) [15]. Then we have filtered the given data so that each entry has a valid Uniprot id, complete primary sequence annotation and 3D information (PDB id). So after this filtration the data size reduced to 1007 interactions with 857 distinct proteins [16]. This database is used for performance evaluation of the proposed algorithm, to identify all possible maximal quasi-cliques from this PPIN, and subsequent pathway analyses.
We have first executed our proposed algorithm for nine different values of \( \upgamma \) starting from 0.9 to 0.1 with an interval of 0.1. In this experiment, we observed that proposed method provides largest quasi-clique with cardinality of 23 for \( \upgamma \) is 0.1. In the second stage, we executed the novel algorithm for finer \( \upgamma \) values in the range (0, 0.1) with an interval of 0.01. We finally observed that algorithm mines largest quasi-clique of size 29 when \( \upgamma \) is 0.07. Considering the value of the \( \upgamma = 0.07 \), we got 49 different maximal quasi-cliques with size ranging from 3 to 29.
For pathway analysis of the clustered proteins, obtained by our proposed algorithm, we have used the web server (http://david.abcc.ncifcrf.gov/tools.jsp), where we first converted proteins ids from Uniprot id to gene id and then analysed all the corresponding genes on KEGG pathway analysis [17]. Our algorithm identifies 46 clusters for KEGG pathway. The Table 1 shows the KEGG pathway of some quasi-cliques along with their respective p-values.
We have compared the performance of the proposed algorithm with the available prior works in this domain. For evaluation of the system performances, the execution time for finding non-redundant disjoint maximal quasi-cliques is considered as one of the key criteria. Two other algorithms, viz., Quick [13] and Cocain [14], are compared with our method on an uniform hardware platform with Intel 2.4 GHz CPU computer having 2 GB internal memory. As discussed before, the performance of our proposed algorithm is optimised for \( \upgamma \) = 0.07. But during comparison of the method with other algorithms, we have considered a wide spectrum of \( \upgamma \) values as 0.01, 0.07, 0.1, 0.5 and 0.9, within the range (0, 1). The detailed results of all methods for all considered \( \upgamma \) values are given in the Table 2. It has been found that our novel algorithm identifies the largest maximal quasi-cliques of cardinality 29 particularly when \( \upgamma \) is 0.07. Overall, our method identifies 49 mutually-exclusive, maximal quasi-cliques from the PPIN and the experiment is completed in 352 s.
The Cocain algorithm works only for \( \upgamma \) values in the range (0.5, 1). Though it takes minimum time for execution in such a specific range, but the major limitation is that the method is not producing significant quasi-cliques for the PPIN. Most of the quasi-cliques are of cardinality 2 or 3 and the total number of quasi-cliques generated by Cocain method ranges in thousands within its allowable \( \upgamma \) range. A detailed comparative analysis of the above mentioned methods is given in the Table 2.
4 Conclusion
In the current work, a new algorithm is presented to find all possible non overlapping non redundant maximal quasi-cliques and also the largest size quasi-clique in the huge PPI networks. Subsequently, we apply this approach on huge PPIN and find important clusters of proteins. We have analysed these protein clusters based KEGG pathway analysis. In this work, we have attempted to cluster interactive human proteins within the PPIN using the developed algorithm and also compared its performance with other available works in this domain. It may be observed that this algorithm is better than other algorithm for identifying non-overlapping, non-redundant maximal quasi-cliques. The performance of the algorithm can be improved when applied over more robust network and also it will be biologically more important.
References
Wagner, A.: How the global structure of protein interaction networks evolves. Proc. Roy. Soc. London Ser. B Biol. Sci. 270, 457–466 (2003)
Du, D., Pardalos, P.M.: Handbook of Combinatorial Optimization. Springer, New York (1998). doi:10.1007/978-1-4613-0303-9
Bomze, I.M., Budinich, M., Pardalos, P.M., et al.: The maximum clique problem. In: Handbook of Combinatorial Optimization, vol. 4, no. 1, pp. 1–74 (1999)
Altaf-Ul-Amin, M., Shinbo, Y., Mihara, K., et al.: Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinform. 7(1), 207 (2006)
Brohee, S., Van Helden, J.: Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinform. 7(1), 488 (2006)
Pereira-Leal, J.B., Enright, A.J., Ouzounis, C.A.: Detection of functional modules from protein interaction networks. Proteins Struct. Funct. Bioinform. 54(1), 49–57 (2003)
Spirin, V., Mirny, L.A.: Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. U.S.A. 100(21), 12123–12128 (2003)
Matsuda, H., Ishihara, T., Hashimoto, A.: Classifying molecular sequences using a linkage graph with their pairwise similarities. Theoret. Comput. Sci. 210(2), 305–325 (1999)
Pei, J., Jiang, D., Zhang, A.: On mining cross-graph quasi-cliques. In: Proceedings of the Eleventh ACM SIGKDD International Conference on Knowledge Discovery in Data Mining, pp. 228–238 (2005)
Jiang, D., Pei, J.: Mining frequent cross-graph quasi-cliques. ACM Trans. Knowl. Discovery Data (TKDD) 2(4), 16 (2009)
Brunato, M., Hoos, Holger H., Battiti, R.: On effectively finding maximal quasi-cliques in graphs. In: Maniezzo, V., Battiti, R., Watson, J.-P. (eds.) LION 2007. LNCS, vol. 5313, pp. 41–55. Springer, Heidelberg (2008). doi:10.1007/978-3-540-92695-5_4
Bhattacharyya, M., Bandyopadhyay, S.: Mining the largest quasi-clique in human protein interactome. In: International Conference on Adaptive and Intelligent Systems, ICAIS 2009, pp. 194–199 (2009)
Liu, G., Wong, L.: Effective pruning techniques for mining quasi-cliques. In: Daelemans, W., Goethals, B., Morik, K. (eds.) ECML PKDD 2008. LNCS, vol. 5212, pp. 33–49. Springer, Heidelberg (2008). doi:10.1007/978-3-540-87481-2_3
Zeng, Z., Wang, J., Zhou, L., et al.: Coherent closed quasi-clique discovery from large dense graph databases. In: Proceedings of the 12th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 797–802 (2006)
Salwinski, L., Miller, C.S., Smith, A.J., et al.: The Database of Interacting Proteins: 2004 Update. Nucleic Acids Res. 32, D449–D451 (2004)
Sriwastava, B.K., Basu, S., Maulik, U., et al.: PPIcons: identification of protein-protein interaction sites in selected organisms. J. Mol. Model. 19(9), 4059–4070 (2013)
Huang, D.W., Sherman, B.T., Lempicki, R.A.: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4(1), 44–57 (2008)
Acknowledgement
This work is partially supported by the CMATER research laboratory of the Computer Science and Engineering Department, Jadavpur University, India, PURSE-II and UPE-II project and Research Award (F.30-31/2016(SA-II)) from UGC, Government of India.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper

Cite this paper
Sriwastava, B.K., Basu, S., Maulik, U. (2017). A Quasi-Clique Mining Algorithm for Analysis of the Human Protein-Protein Interaction Network. In: Shankar, B., Ghosh, K., Mandal, D., Ray, S., Zhang, D., Pal, S. (eds) Pattern Recognition and Machine Intelligence. PReMI 2017. Lecture Notes in Computer Science(), vol 10597. Springer, Cham. https://doi.org/10.1007/978-3-319-69900-4_52
Download citation
DOI: https://doi.org/10.1007/978-3-319-69900-4_52
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-69899-1
Online ISBN: 978-3-319-69900-4
eBook Packages: Computer ScienceComputer Science (R0)
