
Abstract
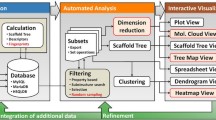
Drug design is very complex and expensive. Finding new active chemical structures is a very important goal. Both experimental and virtual (in silico) screenings can be used to explore chemical space [11][12]. With virtual screening it is possible to reduce the amount of compounds for experimental evaluations. Moreover, when the 3D structure of the target is known, candidate molecules can be put to fit in the target hole in different positions and later cluster these positions in order to find the best to fit. Therefore, we propose a visual tool that couples with Jmol[21] viewer, provides together with a means of visually exploring clustered molecules, an overview of the majority of the data, supporting thus the decision making in the process of new drugs design.
Preview
Unable to display preview. Download preview PDF.
Similar content being viewed by others


References
Wilkinson, L., Anushka, A., Grossman, R.: High-Dimensional Visual Analytics: Interactive Exploration Guided by Pairwise Views of Point Distributions. IEEE Transactions on Visualization and Computer Graphics 12, 1363–1372 (2006)
Saffer, J.D., Burnett, V.L., Chen, G., van der Spek, P.: Visual Analytics in the Pharmaceutical Industry. IEEE Computer Graphics and Applications 24, 10–15 (2004)
Keim, D., Andrienko, G., Fekete, J.D., Kohlhammer Jörn, G., Guy, M.: Visual Analytics: Definition, Process, and Challenges. In: Kerren, A., Stasko, J.T., Fekete, J.-D., North, C. (eds.) Information Visualization. LNCS, vol. 4950, pp. 154–175. Springer, Heidelberg (2008)
Thomas, J.J., Cook, K.A.: A Visual Analytics Agenda. IEEE Comput. Graph. Appl. 26, 10–13 (2006)
Abraham, D.J.: Burgers Medicinal Chemistry and Drug Discovery, 6th edn. John Wiley & Sons/ Wiley-VCH Verlag GmbH & Co. (2003)
Lauss, M., Kriegner, A., Vierlinger, K., Noebammer, C.: Characterization of the drugged human genome. Pharmacogenomics 8, 1063–1073 (2007)
Paolini, G.V., Shapland, R.H.B., van Hoorn, W.P., Mason, J.S., Hopkins, A.L.: Global mapping of pharmacological space. Nat. Biotechnol. 24, 805–815 (2006)
Ojima, I.: Modern Molecular Approaches to Drug Design and Discovery. Acc. Chem. Res. 41, 2–3 (2008)
Baxendale, I.R., Hayward, J.J., Ley, S.V., Tranmer, G.K.: Pharmaceutical Strategy and Innovation: An Academics Perspective. Chem. Med. Chem. 2, 268–288 (2007)
Kubinyi, H.: Drug research: myths, hype and reality. Nat. Rev. Drug Disc. 2, 665–669 (2003)
Ling, X.F.B.: High throughput screening informatics. Comb. Chem. High Throughput Screen 11, 249–257 (2008)
Diller, D.: The synergy between combinatorial chemistry and high-throughput screening. Curr. Opin. Drug Discov. Devel 11, 346–355 (2008)
Soichet, B.K.: Virtual screening of chemical libraries. Nature 432, 862–865 (2004)
Leach, A.R., Shoichet, B.K., Peishoff, C.E.: Prediction of Protein-Ligand Interactions. Docking and Scoring: Successes and Gaps. J. Med. Chem. 49, 5851–5855 (2006)
Warren, G.L., Andrews, C.W., Capelli, A.-M., Clarke, B., LaLonde, J., Lambert, M.H., Lindvall, M., Nevins, N., Semus, S.F., Senger, S., Tedesco, G., Wall, I.D., Woolven, J.M., Peishoff, C.E., Head, M.: A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 49, 5912–5931 (2006)
Sotriffer, C.A., Dramburg, I.: Situ Cross-Docking, To Simultaneously Address Multiple Targets. J. Med. Chem. 48, 3122–3123 (2005)
Peláez, R., Therón, R., García, C.A., López, J.L., Medarde, M.: Design of New Chemoinformatic Tools for the Analysis of Virtual Screening Studies: Application to Tubulin Inhibitors Advances in Soft Computing. In: 2nd International Workshop on Practical Applications of Computational Biology and Bioinformatics (IWPACBB 2008), vol. 49(1), pp. 189–196 (2008)
Carr, R.A.E., Congreve, M., Murray, C.W., Rees, D.C.: Fragment-based lead discovery: leads by design. Drug Disc. Dev. 14, 987–992 (2005)
Irwin, J.J., Shoichet, B.K.: ZINC - A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model 45, 177–182 (2005)
Jain, A.N.: Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 46, 499–511 (2003)
Visualization of the superposed complexes was done with Jmol (2006), http://www.jmol.org and with MarvinBeans 4.1.2, ChemAxon, http://www.chemaxon.com
Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K., Olson, A.J.: Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comp. Chem. 19, 1639–1662 (1998)
Documentation describing the PDB file format, http://www.wwpdb.org/docs.html
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this paper
Cite this paper
García, C.A., Therón, R., Peláez, R., López-Pérez, J.L., Santos-Garcia, G. (2009). Visual Evaluation of Clustered Molecules in the Process of New Drugs Design. In: Butz, A., Fisher, B., Christie, M., Krüger, A., Olivier, P., Therón, R. (eds) Smart Graphics. SG 2009. Lecture Notes in Computer Science, vol 5531. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-02115-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-642-02115-2_1
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-02114-5
Online ISBN: 978-3-642-02115-2
eBook Packages: Computer ScienceComputer Science (R0)