
Abstract
Drug-target interaction (DTI) prediction is a tough task with critical applications in drug repurposing and design scenarios, as it significantly reduces resource consumption and accelerates the drug discovery process. With the proliferation of experimentally measured pharmaceutical data and increasingly complex drug-target interactions, deep DTI approaches are becoming increasingly competitive due to their ability to utilize large-scale data in an end-to-end manner. It is fascinating to consider how to consolidate drug-target pair representations at different granularities to enhance deep DTI models with limited performance. The employed models typically involve solely single granular information and thus lead to a significant lack of features. Motivated by this concern, this study proposes an explainable multi-granularity representation framework for DTI prediction (MultiGranDTI). First, a hierarchical network with constraint is devised to enable the natural conversion of drug representations with different granularities, effectively integrating atomic and substructural information. Second, the 1st-order and 2nd-order sequence information of the target protein is modeled and then encoded together with the spatial information. Subsequently, several convolution layers further extract various levels of protein features. Finally, the drug and protein features are concatenated and fed into regular dense layers for interaction prediction. Comprehensive experiments reveal that MultiGranDTI achieves competitive performance in two types of tasks on four benchmark datasets. Additionally, a visualization case shows that our method is capable of efficiently identifying particular functional groups and substructures in molecules and providing a plausible explanation for the predicted results.
Graphical Abstract
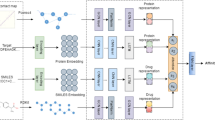
A novel MultiGranDTI model to boost DTI prediction by comprehensively mining the multi-granularity information of compounds and proteins












Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Availability of Data and Materials
The data used to support the findings of this study are available from the corresponding author upon request.
Code Availability
Demonstration, instructions and code for the proposed MultiGranDTI are available at https://github.com/lukcats/MultiGranDTI. For the calculation of protein contact maps, the Pconsc4 can be obtained from https://github.com/ElofssonLab/PconsC4.
References
DeFronzo RA, Inzucchi S, Abdul-Ghani M, Nissen SE (2019) Pioglitazone: the forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diabetes Vasc Dis Res 16(2):133–143
Phillips AN, Cambiano V, Johnson L, Nakagawa F, Homan R, Meyer-Rath G, Rehle T, Tanser F, Moyo S, Shahmanesh M et al (2021) Potential impact and cost-effectiveness of condomless-sex-concentrated prep in kwazulu-natal accounting for drug resistance. J Infect Dis 223(8):1345–1355
Yang X, Niu Z, Liu Y, Song B, Lu W, Zeng L, Zeng X (2023) Modality-dta: Multimodality fusion strategy for drug–target affinity prediction. IEEE/ACM Trans Comput Biol Bioinforma 20(2):1200–1210
Ren Z-H, You Z-H, Zou Q, Yu C-Q, Ma Y-F, Guan Y-J, You H-R, Wang X-F, Pan J (2023) Deepmpf: deep learning framework for predicting drug–target interactions based on multi-modal representation with meta-path semantic analysis. J Transl Med 21(1):1–18
Sajadi SZ, Zare Chahooki MA, Gharaghani S, Abbasi K (2021) Autodti++: deep unsupervised learning for dti prediction by autoencoders. BMC Bioinforma 22(1):1–19
Wang S, Du Z, Ding M, Rodriguez-Paton A, Song T (2022) Kg-dti: a knowledge graph based deep learning method for drug-target interaction predictions and alzheimer’s disease drug repositions. Appl Intell 52(1):846–857
Song D, Chen Y, Min Q, Sun Q, Ye K, Zhou C, Yuan S, Sun Z, Liao J (2019) Similarity-based machine learning support vector machine predictor of drug-drug interactions with improved accuracies. J Clin Pharm Ther 44(2):268–275
An Q, Yu L (2021) A heterogeneous network embedding framework for predicting similarity-based drug-target interactions. Brief Bioinform 22(6):275
Saikia S, Bordoloi M (2019) Molecular docking: challenges, advances and its use in drug discovery perspective. Curr Drug Targets 20(5):501–521
Bagherian M, Sabeti E, Wang K, Sartor MA, Nikolovska-Coleska Z, Najarian K (2021) Machine learning approaches and databases for prediction of drug-target interaction: a survey paper. Brief Bioinform 22(1):247–269
Crampon K, Giorkallos A, Deldossi M, Baud S, Steffenel LA (2022) Machine-learning methods for ligand–protein molecular docking. Drug Discov Today 27(1):151–164
Bonomi M, Vendruscolo M (2019) Determination of protein structural ensembles using cryo-electron microscopy. Curr Opin Struct Biol 56:37–45
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with alphafold. Nature 596(7873):583–589
Sachdev K, Gupta MK (2019) A comprehensive review of feature based methods for drug target interaction prediction. J Biomed Inform 93:103159
He T, Heidemeyer M, Ban F, Cherkasov A, Ester M (2017) Simboost: a read-across approach for predicting drug–target binding affinities using gradient boosting machines. J Cheminformatics 9(1):1–14
Dargan S, Kumar M, Ayyagari MR, Kumar G (2020) A survey of deep learning and its applications: a new paradigm to machine learning. Arch Comput Methods Eng 27:1071–1092
Arús-Pous J, Johansson SV, Prykhodko O, Bjerrum EJ, Tyrchan C, Reymond J-L, Chen H, Engkvist O (2019) Randomized smiles strings improve the quality of molecular generative models. J Cheminformatics 11(1):1–13
Yazdani-Jahromi M, Yousefi N, Tayebi A, Kolanthai E, Neal CJ, Seal S, Garibay OO (2022) Attentionsitedti: an interpretable graph-based model for drug-target interaction prediction using nlp sentence-level relation classification. Brief Bioinform 23(4):272
Chen L, Tan X, Wang D, Zhong F, Liu X, Yang T, Luo X, Chen K, Jiang H, Zheng M (2020) Transformercpi: improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 36(16):4406–4414
Jiang J, Zhang R, Zhao Z, Ma J, Liu Y, Yuan Y, Niu B (2022) Multigran-smiles: multi-granularity smiles learning for molecular property prediction. Bioinformatics 38(19):4573–4580
Nguyen T, Le H, Le T, Venkatesh S (2019) Prediction of drug–target binding affinity using graph neural networks. bioRxiv, 684662
Tsubaki M, Tomii K, Sese J (2019) Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 35(2):309–318
Han S, Fu H, Wu Y, Zhao G, Song Z, Huang F, Zhang Z, Liu S, Zhang W (2023) Himgnn: a novel hierarchical molecular graph representation learning framework for property prediction. Brief Bioinform 24(5):305
Fang Y, Zhang Q, Zhang N, Chen Z, Zhuang X, Shao X, Fan X, Chen H (2023) Knowledge graph-enhanced molecular contrastive learning with functional prompt. Nat Mach Intell 1–12
Öztürk H, Özgür A, Ozkirimli E (2018) Deepdta: deep drug–target binding affinity prediction. Bioinformatics 34(17):821–829
Zheng S, Li Y, Chen S, Xu J, Yang Y (2020) Predicting drug–protein interaction using quasi-visual question answering system. Nat Mach Intell 2(2):134–140
Yuyang W, Wang J, Zhonglin C, Amir BF (2022) Molecular contrastive learning of representations via graph neural networks. Nat Mach Intell 4(3):279–287
Atz K, Grisoni F, Schneider G (2021) Geometric deep learning on molecular representations. Nat Mach Intell 3(12):1023–1032
Lee I, Keum J, Nam H (2019) Deepconv-dti: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput Biol 15(6):1007129
Li C, Yao J, Wei W, Niu Z, Zeng X, Li J, Wang J (2022) Geometry-based molecular generation with deep constrained variational autoencoder. IEEE Transactions on Neural Networks and Learning Systems
Nguyen T, Le H, Quinn TP, Nguyen T, Le TD, Venkatesh S (2021) Graphdta: predicting drug-target binding affinity with graph neural networks. Bioinform 37(8):1140–1147
Kipf TN, Welling M (2017) Semi-supervised classification with graph convolutional networks. In: ICLR 2017
Xu K, Hu W, Leskovec J, Jegelka S (2019) How powerful are graph neural networks? In: ICLR 2019
Jiang M, Li Z, Zhang S, Wang S, Wang X, Yuan Q, Wei Z (2020) Drug–target affinity prediction using graph neural network and contact maps. RSC Adv 10(35):20701–20712
Li F, Zhang Z, Guan J, Zhou S (2022) Effective drug–target interaction prediction with mutual interaction neural network. Bioinformatics 38(14):3582–3589
Wang Q, Huang K, Chandak P, Zitnik M, Gehlenborg N (2022) Extending the nested model for user-centric xai: a design study on gnn-based drug repurposing. IEEE Trans Vis Comput Graph 29(1):1266–1276
Son J, Kim D (2021) Development of a graph convolutional neural network model for efficient prediction of protein-ligand binding affinities. PloS one 16(4):0249404
Ying Z, You J, Morris C, Ren X, Hamilton W, Leskovec J (2018) Hierarchical graph representation learning with differentiable pooling. Adv Neural Inf Process Syst 31
Liu Q, Shu H, Yuan M, Wang G (2022) Fuzzy hierarchical network embedding fusing structural and neighbor information. Inf Sci 603:130–148
Santana CA, Silveira SdA, Moraes JP, Izidoro SC, Melo-Minardi RC, Ribeiro AJ, Tyzack JD, Borkakoti N, Thornton JM (2020) Grasp: a graph-based residue neighborhood strategy to predict binding sites. Bioinformatics 36(Supplement_2):726–734
Michel M, Menéndez Hurtado D, Elofsson A (2019) Pconsc4: fast, accurate and hassle-free contact predictions. Bioinformatics 35(15):2677–2679
Cichonska A, Pahikkala T, Szedmak S, Julkunen H, Airola A, Heinonen M, Aittokallio T, Rousu J (2018) Learning with multiple pairwise kernels for drug bioactivity prediction. Bioinformatics 34(13):509–518
Ying Z, Bourgeois D, You J, Zitnik M, Leskovec J (2019) Gnnexplainer: generating explanations for graph neural networks. Adv Neural Inf Process Syst 32
Yuan H, Yu H, Wang J, Li K, Ji S (2021) On explainability of graph neural networks via subgraph explorations. In: International conference on machine learning, pp 12241–12252
Acknowledgements
This work is supported by the key cooperation project of Chongqing municipal education commission (HZ2021008), partly funded by the State Key Program of National Nature Science Foundation of China (61936001), the Key Research and Development Program of Chongqing (cstc2017zdcy-zdyfx0091), the Key Research and Development Program on AI of Chongqing (cstc2017rgzn-zdyfx0022), and the Doctoral Innovation Talent Program of Chongqing University of Posts and Telecommunications (No.BYJS202301).
Author information
Authors and Affiliations
Contributions
Xu Gong: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Writing Original Draft; Qun Liu: Resources, Methodology, Writing-review and editing, Supervision; Jing He: Software, Data curation, Writing-review and editing; Yike Guo: Conceptualization, Supervision; Guoyin Wang: Supervision, Project administration, Funding acquisition.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no known competingfinancial interests or personal relationships that could haveappeared to influence the work reported in this paper.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
The authors concur the publication by Applied Intelligence.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gong, X., Liu, Q., He, J. et al. MultiGranDTI: an explainable multi-granularity representation framework for drug-target interaction prediction. Appl Intell 55, 107 (2025). https://doi.org/10.1007/s10489-024-05936-7
Accepted:
Published:
DOI: https://doi.org/10.1007/s10489-024-05936-7