
Abstract
The computational determination of binding modes for a ligand into a protein receptor is much more successful than the prediction of relative binding affinities (RBAs) for a set of ligands. Here we consider the binding of a set of 26 synthetic A-CD ligands into the estrogen receptor ERα. We show that the MOE default scoring function (London dG) used to rank the docked poses leads to a negligible correlation with experimental RBAs. However, switching to an energy-based scoring function, using a multiple linear regression to fit experimental RBAs, selecting top-ranked poses and then iteratively repeating this process leads to exponential convergence in 4–7 iterations and a very strong correlation. The method is robust, as shown by various validation tests. This approach may be of general use in improving the quality of predicted binding affinities.











Similar content being viewed by others
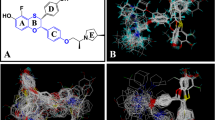
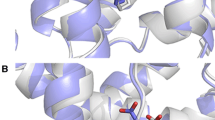
Notes
uDock and auxiliary programs are available free of charge from Dr. Hooman Shadnia’s website at www.shadnia.com (2013).
References
Warren GL, Andrews CW, Capelli A-M, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) A critical assessment of docking programs and scoring functions. J Med Chem 49:5912–5931
Hecht D, Fogel GB (2009) Computational intelligence methods for docking scores. Curr Comp-Aided Drug Des 5:56–68
Englebienne P, Moitessier N (2009) Docking ligands into flexible and solvated macromolecules. 4. Are popular scoring functions accurate for this class of proteins? J Chem Inf Model 49:1568–1580
Englebienne P, Moitessier N (2009) Docking ligands into flexible and solvated macromolecules. 5. Force-field-based prediction of binding affinities of ligands to proteins. J Chem Inf Model 49:2564–2571
Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR (2008) Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br J Pharmacol 153:S7–S26
Corbeil CR, Moitessier N (2009) Docking ligands into flexible and solvated macromolecules. 3. Impact of input ligand conformation, protein flexibility, and water molecules on the accuracy of docking programs. J Chem Inf Model 49:997–1009
Yoon S, Welsh WJ (2004) Identification of a minimal subset of receptor conformations for improved multiple conformation docking and two-step scoring. J Chem Inf Comput Sci 44:88–96
Stjernschantz E, Oostenbrink C (2010) Improved ligand-protein binding affinity predictions using multiple binding modes. Biophys J 98:2682–2691
Labute P, Williams C (2001) Flexible alignment of small molecules. J Med Chem 44:1483–1490
Chan SL, Labute P (2010) Training a scoring function for the alignment of small molecules. J Chem Inf Model 50:1724–1735
Schwarzl SM, Tschopp TB, Smith JC, Fischer S (2002) Can the calculation of ligand binding free energies be improved with continuum solvent electrostatics and an ideal gas entropy correction? J Comput Chem 23:1143–1149
Barone V, Cossi M, Tomasi J (1997) A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J Chem Phys 107:3210–3221
Fornabaio M, Cozzini P, Mozzarelli A, Abraham DJ, Kellogg GE (2003) Simple, intuitive calculations of free energy of binding for protein–ligand complexes. 2. Computational titration and pH effects in molecular models of neuraminidase—inhibitor complexes. J Med Chem 46:4487–4500
Labute P (2009) Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins: Struct Funct Bioinf 75:187–205
Charifson PS, Corkery JJ, Murcko MA, Walters WP (1999) Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem 42:5100–5109
Yang JM, Chen YF, Shen TW, Kristal BS, Hsu DF (2005) Consensus scoring criteria for improving enrichment in virtual screening. J Chem Inf Model 45:1134–1146
Lee FS, Chu Z-T, Bolger MB, Warshel A (1992) Calculations of antibody-antigen interactions: microscopic and semi-microscopic evaluation of the free energies of binding of phosphorylcholine analogs to McPC603. Protein Eng 5:215–228
Aqvist J, Medina C, Samuelsson J-E (1994) A new method for predicting binding affinity in computer-aided drug design. Protein Eng 7:385–391
Hansson T, Aqvist J (1995) Estimation of binding free energies for HIV proteinase inhibitors by molecular dynamics simulations. Protein Eng 8:1137–1144
Aqvist J, Hansson T (1996) On the validity of electrostatic linear response in polar solvents. J Phys Chem 100:9512–9521
Huang S-Y, Zou X (2006) An iterative knowledge-based scoring function to predict protein- ligand interactions: I. Derivation of the interaction potentials. J Comp Chem 27:1866–1874
Jain AN (2006) Scoring functions for protein-ligand docking. Curr Protein Pept Sci 7:407–420
Jain AN (1996) Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput-Aided Mol Design 10:427–440
Jain AN, Harris NL (1995) Park JY quantitative binding site model generation: compass applied to multiple chemotypes targeting the 5-HT1A receptor. J Med Chem 38:1295–1308
Durrant JD, McCammon JA (2010) NNscore: a neural-network-based scoring function for the characterization of protein-ligand complexes. J Chem Inf Model 50:1865–1871
Durrant JD, McCammon JA (2011) NNScore 2.0: a neural network receptor-ligand scoring function. J Chem Inf Model 51:2897–2903
Deng W, Breneman C, Embrechts MJ (2004) Predicting protein-ligand binding affinities using novel geometrical descriptors and machine-learning methods. J Chem Inf Comput Sci 44:699–703
Ballester PJ, Mitchell JB (2010) A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 26:1169–1175
Kinnings SL, Liu N, Tonge PJ, Jackson RM, Xie L, Bourne PE (2011) A machine learning based method to improve docking scoring functions and its application to drug repurposing. J Chem Inf Model 51:408–419
Martin E, Sullivan DC (2008) AutoShim: empirically corrected scoring functions for quantitative docking with a crystal structure and IC50 training data. J Chem Inf Model 48:861–872
Martin E, Sullivan DC (2008) Surrogate autoshim: predocking into a universal ensemble kinase receptor for three dimensional activity prediction, very quickly, without a crystal structure. J Chem Inf Model 48:873–881
Molecular Operating Environment (MOE) (2012) 10; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2012
Asim M, El-Safiti M, Qian Y, Choueiri C, Salari S, Cheng J, Shadnia H, Bal M, Pratt MAC, Carlson KE, Katzenellenbogen JA, Wright JS, Durst T (2009) Deconstructing estradiol: removal of B-ring generates compounds which are potent and subtype-selective estrogen receptor agonists. Bioorg Med Chem Lett 19:1250–1253; [Erratum to document cited in (2009) Bioorg Med Chem Lett 19: 2605]
Wright JS, Shadnia H, Anderson JM, Durst T, Asim M, El-Safiti M, Choueiri C, Pratt MAC, Ruddy SC, Lau R, Carlson KE, Katzenellenbogen JA, O’Brien PJ, Wan L (2011) A-CD Estrogens: i. Substituent effects, hormone potency, and receptor subtype selectivity in a new family of flexible estrogenic compounds. J Med Chem 54:433–448
Merz KM (2010) Limits of free energy computation for protein-ligand combinations. J Chem Theory Comput 6:1769–1776
Gaussian 03, revision C.02; Gaussian, Inc.: Wallingford, CT, 2003
Halgren TA (1996) Merck molecular force field. I. Basis, form, scope, parameterization and performance of MMFF94. J Comput Chem 17:490–517
Acknowledgments
This work was funded by the Canadian Breast Cancer Foundation (Ontario Region) grants to J. S. Wright and T. Durst. We are grateful to the CBCF for support for this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Wright, J.S., Anderson, J.M., Shadnia, H. et al. Experimental versus predicted affinities for ligand binding to estrogen receptor: iterative selection and rescoring of docked poses systematically improves the correlation. J Comput Aided Mol Des 27, 707–721 (2013). https://doi.org/10.1007/s10822-013-9670-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-013-9670-6