
Abstract
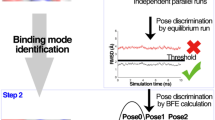
The binding mode prediction is of great importance to structure-based drug design. The discrimination of various binding poses of ligand generated by docking is a great challenge not only to docking score functions but also to the relatively expensive free energy calculation methods. Here we systematically analyzed the stability of various ligand poses under molecular dynamics (MD) simulation. First, a data set of 120 complexes was built based on the typical physicochemical properties of drug-like ligands. Three potential binding poses (one correct pose and two decoys) were selected for each ligand from self-docking in addition to the experimental pose. Then, five independent MD simulations for each pose were performed with different initial velocities for the statistical analysis. Finally, the stabilities of ligand poses under MD were evaluated and compared with the native one from crystal structure. We found that about 94% of the native poses were maintained stable during the simulations, which suggests that MD simulations are accurate enough to judge most experimental binding poses as stable properly. Interestingly, incorrect decoy poses were maintained much less and 38–44% of decoys could be excluded just by performing equilibrium MD simulations, though 56–62% of decoys were stable. The computationally-heavy binding free energy calculation can be performed only for these survived poses.






Similar content being viewed by others


References
Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL (2010) How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 9:203–214
Bajorath J (2002) Integration of virtual and high-throughput screening. Nat Rev Drug Discov 1:882–894
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3:935–949
Reddy AS, Pati SP, Kumar PP, Pradeep HN, Sastry GN (2007) Virtual screening in drug discovery—a computational perspective. Curr Protein Pept Sci 8:329–351
Leach AR, Shoichet BK, Peishoff CE (2006) Prediction of protein–ligand interactions. Docking and scoring: successes and gaps. J Med Chem 49:5851–5855
Warren GL, Andrews CW, Capelli A-M, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) A critical assessment of docking programs and scoring functions. J Med Chem 49:5912–5931
Kroemer RT (2007) Structure-based drug design: docking and scoring. Curr Protein Pept Sci 8:312–328
Dunbar JB, Smith RD, Damm-Ganamet KL, Ahmed A, Esposito EX, Delproposto J, Chinnaswamy K, Kang Y-N, Kubish G, Gestwicki JE, Stuckey JA, Carlson HA (2013) CSAR data set release 2012: ligands, affinities, complexes, and docking decoys. J Chem Inf Model 53:1842–1852
Huang N, Shoichet BK, Irwin JJ (2006) Benchmarking sets for molecular docking. J Med Chem 49:6789–6801
Li Y, Liu Z, Li J, Han L, Liu J, Zhao Z, Wang R (2014) Comparative assessment of scoring functions on an updated benchmark: 1. compilation of the test set. J Chem Inf Model 54:1700–1716
Lagarde N, Zagury J-F, Montes M (2015) Benchmarking data sets for the evaluation of virtual ligand screening methods: review and perspectives. J Chem Inf Model 55:1297–1307
Halperin I, Ma B, Wolfson H, Nussinov R (2002) Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins 47:409–443
Cheng T, Li X, Li Y, Liu Z, Wang R (2009) Comparative assessment of scoring functions on a diverse test set. J Chem Inf Model 49:1079–1093
Li Y, Han L, Liu Z, Wang R (2014) Comparative assessment of scoring functions on an updated benchmark: 2. evaluation methods and general results. J Chem Inf Model 54:1717–1736
Hamza A, Wei N-N, Zhan C-G (2012) Ligand-based virtual screening approach using a new scoring function. J Chem Inf Model 52:963–974
Nabuurs SB, Wagener M, de Vlieg J (2007) A flexible approach to induced fit docking. J Med Chem 50:6507–6518
Sun H, Li Y, Shen M, Tian S, Xu L, Pan P, Guan Y, Hou T (2014) Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys Chem Chem Phys 16:22035–22045
Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the MM/PBSA and MM/GBSA Methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 51:69–82
Greenidge PA, Kramer C, Mozziconacci J-C, Sherman W (2014) Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. J Chem Inf Model 54:2697–2717
Thompson DC, Humblet C, Joseph-McCarthy D (2008) Investigation of MM-PBSA rescoring of docking poses. J Chem Inf Model 48:1081–1091
Cao R, Huang N, Wang Y (2014) Evaluation and application of MD-PB/SA in structure-based hierarchical virtual screening. J Chem Inf Model 54:1987–1996
Rastelli G, Del Rio A, Degliesposti G, Sgobba M (2010) Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem 31:797–810
Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J Comput Chem 32:866–877
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10:449–461
Mobley DL, Dill KA (2009) Binding of small-molecule ligands to proteins: “what you see” is not always “what you get”. Structure 17:489–498
Clark AJ, Tiwary P, Borrelli K, Feng S, Miller EB, Abel R, Friesner RA, Berne BJ (2016) Prediction of protein–ligand binding poses via a combination of induced fit docking and metadynamics simulations. J Chem Theory Comput 12:2990–2998
Kellenberger E, Muller P, Schalon C, Bret G, Foata N, Rognan D (2006) sc-PDB: an annotated database of druggable binding sites from the Protein Data Bank. J Chem Inf Model 46:717–727
Desaphy J, Bret G, Rognan D, Kellenberger E (2015) sc-PDB: a 3D-database of ligandable binding sites–10 years on. Nucleic Acids Res 43:D399–D404
Hu L, Benson ML, Smith RD, Lerner MG, Carlson HA (2005) Binding MOAD (mother of all databases). Proteins Struct Funct Bioinform 60:333–340
Ahmed A, Smith RD, Clark JJ, Dunbar JB, Carlson HA (2015) Recent improvements to Binding MOAD: a resource for protein–ligand binding affinities and structures. Nucleic Acids Res 43:D465–D469
Kerns EH, Di L (2008) Drug-like properties concepts, structure design and methods: from ADME to toxicity optimization. Academic Press, Boston
Michalsky E, Dunkel M, Goede A, Preissner R (2005) SuperLigands—a database of ligand structures derived from the Protein Data Bank. BMC Bioinform 6:122
Leeson PD, Springthorpe B (2007) The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov 6:881–890
Leeson PD, Young RJ (2015) Molecular property design: does everyone get it? ACS Med Chem Lett 6:722–725
Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL (2012) Quantifying the chemical beauty of drugs. Nat Chem 4:90–98
Warren GL, Do TD, Kelley BP, Nicholls A, Warren SD (2012) Essential considerations for using protein–ligand structures in drug discovery. Drug Discov Today 17:1270–1281
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Dyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Chemical Computing Group Inc. (2014) Molecular Operating Environment (MOE), 2014.09. 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09 Revision, vol C 01. Gaussian Inc, Wallingford CT
Walker RC, Crowley MF, Case DA (2008) The implementation of a fast and accurate QM/MM potential method in Amber. J Comput Chem 29:1019–1031
Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham, III TE, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Luchko T, Luo R, Madej B, Merz KM, Monard G, Needham P, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Salomon-Ferrer R, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM (2015) AMBER 2015. University of California, San Francisco
Bradbrook GM, Gleichmann T, Harrop SJ, Habash J, Raftery J, Kalb (Gilboa) J, Yariv J, Hillier IH, Helliwell JR (1998) X-Ray and molecular dynamics studies of concanavalin-A glucoside and mannoside complexes Relating structure to thermodynamics of binding. J Chem Soc Faraday Trans 94:1603–1611
Li P, Merz KM (2014) Taking into account the ion-induced dipole interaction in the nonbonded model of ions. J Chem Theory Comput 10:289–297
Sindhikara DJ, Kim S, Voter AF, Roitberg AE (2009) Bad seeds sprout perilous dynamics: stochastic thermostat induced trajectory synchronization in biomolecules. J Chem Theory Comput 5:1624–1631
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34:D668–D672
Chipot C, Pohorille A (2007) Free energy calculations: theory and applications in chemistry and biology. Springer, Berlin
Kovalenko A, Hirata F (1999) Self-consistent description of a metal-water interface by the Kohn–Sham density functional theory and the three-dimensional reference interaction site model. J Chem Phys 110:10095
Essex JW, Jorgensen WL (1995) An empirical boundary potential for water droplet simulations. J Comput Chem 16:951–997
Woo H-J, Dinner AR, Roux B (2004) Grand canonical Monte Carlo simulations of water in protein environments. J Chem Phys 121:6392
Ahmed A, Sandler SI (2013) Hydration free energies of multifunctional nitroaromatic compounds. J Chem Theory Comput 9:2774–2785
Acknowledgements
The authors are grateful to Dr. Satoshi Endo and Dr. Toshimasa Tanaka for careful reading of the manuscript and valuable suggestions and comments. The computational resources are supported by the K computer of the RIKEN Advanced Institute for Computational Science through the HPCI System Research project (Project ID: hp120013) and the TSUBAME Grid Cluster at the Global Scientific Information and Computing Center of Tokyo Institute of Technology, supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) Open Advanced Research Facilities Initiative (Project ID: 15IBD).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
This material is available free of charge via the Internet at http://rd.springer.com/journal/10822.
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, K., Watanabe, E. & Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J Comput Aided Mol Des 31, 201–211 (2017). https://doi.org/10.1007/s10822-016-0005-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-016-0005-2