
Abstract
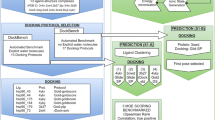
Molecular docking is a powerful tool in the field of computer-aided molecular design. In particular, it is the technique of choice for the prediction of a ligand pose within its target binding site. A multitude of docking methods is available nowadays, whose performance may vary depending on the data set. Therefore, some non-trivial choices should be made before starting a docking simulation. In the same framework, the selection of the target structure to use could be challenging, since the number of available experimental structures is increasing. Both issues have been explored within this work. The pose prediction of a pool of 36 compounds provided by D3R Grand Challenge 2 organizers was preceded by a pipeline to choose the best protein/docking-method couple for each blind ligand. An integrated benchmark approach including ligand shape comparison and cross-docking evaluations was implemented inside our DockBench software. The results are encouraging and show that bringing attention to the choice of the docking simulation fundamental components improves the results of the binding mode predictions.







Similar content being viewed by others

References
Talele TT, Khedkar SA, Rigby AC (2010) Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr Top Med Chem 10:127–141
Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE (1982) A geometric approach to macromolecule–ligand interactions. J Mol Biol 161:269–288
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
McGovern SL, Shoichet BK (2003) Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J Med Chem 46:2895–2907
Directory of in silico Drug Design tools. http://www.click2drug.org/. Accessed 1 Jun 2017
Claudel T, Staels B, Kuipers F (2005) The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol 25:2020–2030
Salmaso V, Sturlese M, Cuzzolin A, Moro S (2016) DockBench as docking selector tool: the lesson learned from D3R Grand Challenge 2015. J Comput Aided Mol Des 30:773–789
Gathiaka S, Liu S, Chiu M et al (2016) D3R grand challenge 2015: evaluation of protein-ligand pose and affinity predictions. J Comput Aided Mol Des 30:651–668
Cuzzolin A, Sturlese M, Malvacio I, Ciancetta A, Moro S (2015) DockBench: an integrated informatic platform bridging the gap between the robust validation of docking protocols and virtual screening simulations. Molecules 20:9977–9993
Kollman PA, Massova I, Reyes C et al (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33:889–897
Harvey MJ, Giupponi G, Fabritiis GD (2009) ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput 5:1632–1639
Chemical Computing Group (CCG) Inc (2016) Molecular operating environment (MOE)
Labute P (2009) Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 75:187–205
Schrödinger (2017) Schrödinger release 2017-1: LigPrep. New York, NY
RDKit: Open-source cheminformatics. http://www.rdkit.org. Accessed 28 May 2017
Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 50:572–584
Hawkins PCD, Skillman AG, Nicholls A (2007) Comparison of shape-matching and docking as virtual screening tools. J Med Chem 50:74–82
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Friesner RA, Banks JL, Murphy RB et al (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47:1750–1759
Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD (2003) Improved protein-ligand docking using GOLD. Proteins 52:609–623
Korb O, Stützle T, Exner TE (2007) An ant colony optimization approach to flexible protein–ligand docking. Swarm Intell 1:115–134
Korb O, Stützle T, Exner TE (2009) Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inf Model 49:84–96
Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P, Barril X, Hubbard RE, Morley SD (2014) rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput Biol 10:e1003571
Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA (2014) AMBER 14. University of California, San Francisco
Gasteiger J, Marsili M (1980) Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 36:3219–3228
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J Chem Phys 98:10089
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577
Onufriev A, Bashford D, Case DA (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55:383–394
Weiser J, Shenkin PS, Still CW (1999) Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem
Downes M, Verdecia MA, Roecker AJ et al (2003) A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol Cell 11:1079–1092
Soisson SM, Parthasarathy G, Adams AD, Sahoo S, Sitlani A, Sparrow C, Cui J, Becker JW (2008) Identification of a potent synthetic FXR agonist with an unexpected mode of binding and activation. Proc Natl Acad Sci USA 105:5337–5342
Akwabi-Ameyaw A, Bass JY, Caldwell RD et al (2008) Conformationally constrained farnesoid X receptor (FXR) agonists: naphthoic acid-based analogs of GW 4064. Bioorg Med Chem Lett 18:4339–4343
Flatt B, Martin R, Wang T-L et al (2009) Discovery of XL335 (WAY-362450), a highly potent, selective, and orally active agonist of the farnesoid X receptor (FXR). J Med Chem 52:904–907
Feng S, Yang M, Zhang Z et al (2009) Identification of an N-oxide pyridine GW4064 analog as a potent FXR agonist. Bioorg Med Chem Lett 19:2595–2598
Bass JY, Caldwell RD, Caravella JA et al (2009) Substituted isoxazole analogs of farnesoid X receptor (FXR) agonist GW4064. Bioorg Med Chem Lett 19:2969–2973
Akwabi-Ameyaw A, Bass JY, Caldwell RD et al (2009) FXR agonist activity of conformationally constrained analogs of GW 4064. Bioorg Med Chem Lett 19:4733–4739
Lundquist JT, Harnish DC, Kim CY et al (2010) Improvement of physiochemical properties of the tetrahydroazepinoindole series of farnesoid X receptor (FXR) agonists: beneficial modulation of lipids in primates. J Med Chem 53:1774–1787
Richter HGF, Benson GM, Blum D et al (2011) Discovery of novel and orally active FXR agonists for the potential treatment of dyslipidemia & diabetes. Bioorg Med Chem Lett 21:191–194
Richter HGF, Benson GM, Bleicher KH et al (2011) Optimization of a novel class of benzimidazole-based farnesoid X receptor (FXR) agonists to improve physicochemical and ADME properties. Bioorg Med Chem Lett 21:1134–1140
Bass JY, Caravella JA, Chen L et al (2011) Conformationally constrained farnesoid X receptor (FXR) agonists: heteroaryl replacements of the naphthalene. Bioorg Med Chem Lett 21:1206–1213
Akwabi-Ameyaw A, Caravella JA, Chen L et al (2011) Conformationally constrained farnesoid X receptor (FXR) agonists: alternative replacements of the stilbene. Bioorg Med Chem Lett 21:6154–6160
Xu X, Xu X, Liu P, Zhu Z, Chen J, Fu H, Chen L, Hu L, Shen X (2015) Structural basis for SMALL MOLECULE NDB (N-benzyl-N-(3-(tert-butyl)-4-hydroxyphenyl)-2,6-dichloro-4-(dimethylamino) benzamide) as a selective antagonist of farnesoid X receptor α (FXRα) in stabilizing the homodimerization of the receptor. J Biol Chem 290:19888–19899
Kudlinzki D, Merk D, Linhard VL, Saxena K, Sreeramulu S, Nilsson E, Dekker N, Wissler L, Bamberg K, Schubert-Zsilavecz M, Schwalbe H (2015) FXR with CDCA and NCoA-2 peptide. http://www.rcsb.org/pdb/explore.do?structureId=4qe6. Accessed 23 Aug 2017
Jin L, Feng X, Rong H et al (2013) The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat Commun 4:1937
Pedregosa F, Varoquaux G, Gramfort A et al (2011) Scikit-learn: machine learning in python. J Mach Learn Res 12:2825–2830
Ester M, Kriegel H-P, Sander J, Xu X (1996) A density-based algorithm for discovering clusters in large spatial databases with noise. In: Proceedings of the 2nd international conference on knowledge discovery and data mining. Portland, OR, pp 226–231
Acknowledgements
MMS lab is also very grateful to Chemical Computing Group, OpenEye, and Acellera for the scientific and technical partnership. S.M. participates in the European COST Action CM1207 (GLISTEN). Finally, MMS lab is extremely grateful to the organizers of the D3R Grand Challenge for the perfect organization of the event.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Salmaso, V., Sturlese, M., Cuzzolin, A. et al. Combining self- and cross-docking as benchmark tools: the performance of DockBench in the D3R Grand Challenge 2. J Comput Aided Mol Des 32, 251–264 (2018). https://doi.org/10.1007/s10822-017-0051-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-017-0051-4