
Abstract
In epigenome-wide association studies (EWAS), the mixed methylation expression caused by the combination of different cell types may lead the researchers to find the false methylation site related to the phenotype of interest. To correct the EWAS false discovery, some non-reference models based on sparse principal component analysis (sparse PCA) have been proposed. These models assume that all methylation sites have the same priori probability in each PC load. However, it is known that there already has gene network structure corresponding to the methylation site. How to integrate this genome network knowledge into the sparse PCA models to enhance the performance of existing models is an open research problem. We introduce GN-ReFAEWAS, a non-reference analysis model which integrates the prior gene network structure into the PCA framework to control the false discovery in EWAS. We used one simulated data set, three real data sets, and three additional tests for experiments and compared with four existing models. Experimental results show that the GN-ReFAEWAS model is better than the existing model by 2–90% in the indicators of sensitivity, specificity, genomic control factor λ, and correlation coefficient factor cov with known cell phenotype ratio.
Graphical abstract














Similar content being viewed by others

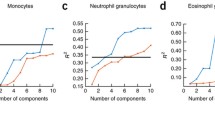
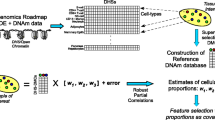
Data Availability
A sample code in R language is available at: https://github.com/mr1528126360/GNReFAEWAS.
References
Flanagan JM (2015) Epigenome-wide association studies (EWAS): past, present, and future. Cancer Epigenetics: Springer:51–63
Verma M (2012) Epigenome-wide association studies (EWAS) in cancer. Curr Genomics 13(4):308–313
Michels KB et al (2013) Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods 10(10):949
Braun KV et al (2017) Epigenome-wide association study (EWAS) on lipids: the Rotterdam Study. Clin Epigenetics 9(1):1–11
Johansson A, Flanagan JM (2017) Epigenome-wide association studies for breast cancer risk and risk factors. Trends Cancer Res 12:19
Shenker NS et al (2013) Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet 22(5):843–851
Nustad HE et al (2022) Modeling dependency structures in 450k DNA methylation data. Bioinformatics 38(4):885–891
Ghosh M, Sen S, Sarkar R, Maulik U (2021) Quantum squirrel inspired algorithm for gene selection in methylation and expression data of prostate cancer. Appl Soft Comput 105:107221
Murphy TM, Mill J (2014) Epigenetics in health and disease: heralding the EWAS era. Lancet 383(9933):1952–1954
Li M et al (2019) EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res 47(D1):D983–D988
Jaffe AE, Irizarry RA (2014) Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 15(2):1–9
Zou J, Lippert C, Heckerman D, Aryee M, Listgarten J (2014) Epigenome-wide association studies without the need for cell-type composition. Nat Methods 11(3):309–311
Naeem H et al (2014) Reducing the risk of false discovery enabling identification of biologically significant genome-wide methylation status using the HumanMethylation450 array. BMC Genomics 15(1):51
Patel CJ, Bhattacharya J, Butte AJ (2010) An environment-wide association study (EWAS) on type 2 diabetes mellitus. PLoS ONE 5(5):e10746
Houseman EA et al (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13(1):86
Graw S, Henn R, Thompson JA, Koestler DC (2019) pwrEWAS: a user-friendly tool for comprehensive power estimation for epigenome wide association studies (EWAS). BMC Bioinformatics 20(1):218
Houseman EA, Kelsey KT, Wiencke JK, Marsit CJ (2015) Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinformatics 16(1):1–16
Yang B, Bao W, Wang J (2022) Active disease-related compound identification based on capsule network. Brief Bioinform 23(1):bbab462
Bao W et al (2017) Mutli-features prediction of protein translational modification sites. IEEE/ACM Trans Comput Biol Bioinformatics 15(5):1453–1460
Bao W, Wang D, Chen Y (2016) Classification of protein structure classes on flexible neutral tree. IEEE/ACM Trans Comput Biol Bioinformatics 14(5):1122–1133
Zheng X et al (2014) MethylPurify: tumor purity deconvolution and differential methylation detection from single tumor DNA methylomes. Genome Biol 15(7):1–13
Houseman EA, Molitor J, Marsit CJ (2014) Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 30(10):1431–1439
Newman AM et al (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12(5):453–457
Yoshihara K et al (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4(1):1–11
Koestler DC et al (2013) Blood-based profiles of DNA methylation predict the underlying distribution of cell types: a validation analysis. Epigenetics 8(8):816–826
Accomando WP, Wiencke JK, Houseman EA, Nelson HH, Kelsey KT (2014) Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol 15(3):R50
Teschendorff AE, Breeze CE, Zheng SC, Beck S (2017) A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinformatics 18(1):105
Reinius LE et al (2012) Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE 7(7):e41361
Koestler DC et al (2016) Improving cell mixture deconvolution by identifying optimal DNA methylation libraries (IDOL). BMC Bioinformatics 17(1):120
Olova N et al (2018) Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. Genome Biol 19(1):1–19
Zhang Y et al (2019) Factors affecting differential methylation of DNA promoters in arsenic-exposed populations. Biol Trace Elem Res 189(2):437–446
Dagar V et al (2018) Genetic variation affecting DNA methylation and the human imprinting disorder, Beckwith-Wiedemann syndrome. Clin Epigenetics 10(1):114
Houseman EA, Kile ML, Christiani DC, Ince TA, Kelsey KT, Marsit CJ (2016) Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics 17(1):259
McGregor K et al (2016) An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies. Genome Biol 17(1):84
Rahmani E et al (2016) Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods 13(5):443
Li S et al (2013) An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics 14(5):1–9 (BioMed Central)
Journée M, Nesterov Y, Richtárik P, Sepulchre R (2010) Generalized power method for sparse principal component analysis. Journal of Machine Learning Research 11(2):517–553
Yuan X-T, Zhang T (2013) Truncated power method for sparse eigenvalue problems. J Mach Learn Res 14(Apr):899–925
Liu W, Zhang H, Tao D, Wang Y, Lu K (2016) Large-scale paralleled sparse principal component analysis. Multimed Tools Appl 75(3):1481–1493
Hoffmann R, Valencia A (2004) A gene network for navigating the literature. Nat Genet 36(7):664–664
Bartlett TE, Olhede SC, Zaikin A (2014) A DNA methylation network interaction measure, and detection of network oncomarkers. PLoS ONE 9(1):e84573
van Eijk KR et al (2012) Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics 13(1):636
Kim K, Sun H (2019) Incorporating genetic networks into case-control association studies with high-dimensional DNA methylation data. BMC Bioinformatics 20(1):510
Saeliw T et al (2018) Integrated genome-wide Alu methylation and transcriptome profiling analyses reveal novel epigenetic regulatory networks associated with autism spectrum disorder. Mol Autism 9(1):27
Jones A et al (2013) Role of DNA methylation and epigenetic silencing of HAND2 in endometrial cancer development. PLoS Med 10(11):e1001551
Jiao Y, Widschwendter M, Teschendorff AE (2014) A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control. Bioinformatics 30(16):2360–2366
Mignone P, Pio G, Džeroski S, Ceci M (2020) Multi-task learning for the simultaneous reconstruction of the human and mouse gene regulatory networks. Sci Rep 10(1):1–15
Hyvärinen A, Oja E (2000) Independent component analysis: algorithms and applications. Neural Netw 13(4–5):411–430
Leek J, Storey J (2007) Bioconductor’s sva package. Dim (svadat) 1(1000):20
Houseman EA et al (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13(1):1–16
Balding DJ (2006) A tutorial on statistical methods for population association studies. Nat Rev Genet 7(10):781–791
Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55(4):997–1004
Zhou Y et al (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10(1):1–10
Kuleshov MV et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44(W1):W90–W97
Acknowledgements
We are very grateful to the valuable comments by the anonymous reviewers.
Funding
This work is supported by the Macau Science and Technology Development Funds Grands No. 0056/2020/AFJ from the Macau Special Administrative Region of the People’s Republic of China, Key Project for University of Educational Commission of Guangdong Province of China Funds (Natural, Grant No. 2019GZDXM005), and Research and Demonstration of East and West Cerebral Infarction Recurrence Prediction Model Construction and Early Warning System Development Based on Multi-omics, Science and Technology Project of Guizhou Province, Project Number: Qian Ke He Support [2021] General 446.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Miao, R., Dang, Q., Cai, J. et al. Sparse principal component analysis based on genome network for correcting cell type heterogeneity in epigenome-wide association studies. Med Biol Eng Comput 60, 2601–2618 (2022). https://doi.org/10.1007/s11517-022-02599-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11517-022-02599-9