
Abstract
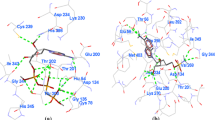
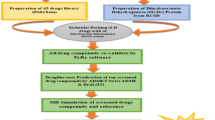
S-adenosyl-l-homocysteine hydrolase of Plasmodium falciparum (PfSAHH) has been reported as a potential drug target against malaria. A series of aristeromycin derivatives and analogs were designed and tested for inhibition of PfSAHH. 2-Fluoroaristeromycin has been reported as a potential inhibitor of PfSAHH. Here, we have performed the molecular dynamics simulation study of 2-Fluoroaristeromycin with PfSAHH with 15-ns simulation time to evaluate the dynamic perturbation of inhibitor in the binding site of PfSAHH in docked complex. This indicates that the complex structure of PfSAHH-2-Fluoroaristeromycin is stable after 10 ns of simulation. MD results indicate that Leu53, His54, Thr56, Glu58, Cys59, Asp134, Glu200, Lys230, Leu389, Leu392, Gly397, Hip398, Met403, and Phe407 are the key residues in the binding pocket of PfSAHH that interacts with the inhibitor 2-Fluoroaristeromycin. Earlier studies have reported Cys59 of PfSAHH as a selective residue to design potential and specific inhibitor of PfSAHH. Simulation study also indicates the role of Cys59 in binding interaction with inhibitor. The MD simulation of PfSAHH-2-Fluoroaristeromycin complex reveals the stable nature of docking interaction. The result provides a set of guidelines for the rational design of potential inhibitors of PfSAHH.





Similar content being viewed by others
References
Andersen HC (1983) Rattle: a “velocity” version of the shake algorithm for molecular dynamics calculations. J Comput Phys 52(1):24–34
Ando T, Iwata M, Zulfiqar F, Miyamoto T, Nakanishi M, Kitade Y (2008a) Synthesis of 2-modified aristeromycins and their analogs as potent inhibitors against Plasmodium falciparum S-adenosyl-l-homocysteine hydrolase. Bioorg Med Chem 16(7):3809–3815
Ando T, Kojima K, Chahota P, Kozaki A, Milind ND, Kitade Y (2008b) Synthesis of 4′-modified noraristeromycins to clarify the effect of the 4′-hydroxyl groups for inhibitory activity against S-adenosyl-l-homocysteine hydrolase. Bioorg Med Chem Lett 18(8):2615–2618
Bowers KJ, Chow E, Xu H, Dror R O, Eastwood M P, Gregerson BA, Klepeis JL, Kolossvary I, Moraes M A, Sacerdoti FD, Salmon JK, Shan Y, Shaw D E (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters. In: Proc. ACM/IEEE conf. on supercomputing (Tampa, FL, 2006)
Bujnicki JM, Prigge ST, Caridha D, Chiang PK (2003) Structure, evolution, and inhibitor interaction of S-adenosyl-l-homocysteine hydrolase from Plasmodium falciparum. Proteins 52(4):624–632
Cai S, Li QS, Borchardt RT, Kuczera K, Schowen RL (2007) The antiviral drug ribavirin is a selective inhibitor of S-adenosyl-l-homocysteine hydrolase from Trypanosoma cruzi. Bioorg Med Chem 15(23):7281–7287
Carugo O (2007) Statistical validation of the root-mean-square-distance, a measure of protein structural proximity. Protein Eng Des Sel 20(1):33–37
Carugo O, Pongor S (2001) A normalized root-mean-square distance for comparing protein three-dimensional structures. Protein Sci 10(7):1470–1473
Chiang PK (1998) Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol Ther 77(2):115–134
Das SR, Schneller SW, Balzarini J, De Clercq E (2002) A mercapto analogue of 5′-noraristeromycin. Bioorg Med Chem 10(2):457–460
Hudec R, Hamada K, Mikoshiba K (2013) A fluorescence-based assay for the measurement of S-adenosylhomocysteine hydrolase activity in biological samples. Anal Biochem 433(2):95–101
Ito M, Hamano T, Komatsu T, Asamitsu K, Yamakawa T, Okamoto T (2014) A novel IKKα inhibitor, noraristeromycin, blocks the chronic inflammation associated with collagen-induced arthritis in mice. Mod Rheumatol 24(5):775–780
Kaminski GA, Friesner RA (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem 105:6474–6487
Kitade Y, Kozaki A, Miwa T, Nakanishi M (2002) Synthesis of base-modified noraristeromycin derivatives and their inhibitory activity against human and Plasmodium falciparum recombinant S-adenosyl-l-homocysteine hydrolase. Tetrahedron 58:1271–1277
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791
Nakanishi M (2007) S-adenosyl-l-homocysteine hydrolase as an attractive target for antimicrobial drugs. Yakugaku Zasshi 127(6):977–982
Seley KL, Schneller SW, Rattendi D, Bacchi CJ (1997) (+)-7-Deaza-5′-noraristeromycin as an anti-trypanosomal agent. J Med Chem 40(4):622–624
Shaw DE (2008) Research desmond molecular dynamics system, version 3.1. New York
Singh DB, Gupta MK, Singh DV, Singh SK, Misra K (2013) Docking and in silico ADMET studies of noraristeromycin, curcumin and its derivatives with Plasmodium falciparum SAH hydrolase: a molecular drug target against malaria. Interdiscip Sci 5(1):1–12
Tanaka N, Umeda T, Kusakabe Y, Nakanishi M, Kitade Y, Nakamura KT (2013) Structural biology for developing antimalarial compounds. Yakugaku Zasshi 133(5):527–537
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321
Wang Y, Kavran JM, Chen Z, Karukurichi KR, Leahy DJ, Cole PA (2014) Regulation of S-adenosylhomocysteine hydrolase by lysine acetylation. J Biol Chem 289(45):31361–31372
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Singh, D.B., Dwivedi, S. Docking and molecular dynamics simulation study of inhibitor 2-Fluoroaristeromycin with anti-malarial drug target PfSAHH. Netw Model Anal Health Inform Bioinforma 5, 16 (2016). https://doi.org/10.1007/s13721-016-0124-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-016-0124-7
Keywords
Profiles
- Dev Bukhsh Singh View author profile