
Abstract
Solubility parameter based methods have long been a valuable tool for solvent formulation and selection. Of these methods, the MOdified Separation of Cohesive Energy Density (MOSCED) has recently been shown to correlate well the equilibrium solubility of multifunctional non-electrolyte solids. However, before it can be applied to a novel solute, a limited amount of reference solubility data is required to regress the necessary MOSCED parameters. Here we demonstrate for the solutes methylparaben, ethylparaben, propylparaben, butylparaben, lidocaine and ephedrine how conventional molecular simulation free energy calculations or electronic structure calculations in a continuum solvent, here the SMD or SM8 solvation model, can instead be used to generate the necessary reference data, resulting in a predictive flavor of MOSCED. Adopting the melting point temperature and enthalpy of fusion of these compounds from experiment, we are able to predict equilibrium solubilities. We find the method is able to well correlate the (mole fraction) equilibrium solubility in non-aqueous solvents over four orders of magnitude with good quantitative agreement.







Similar content being viewed by others

Notes
Recall that \(\ln \frac{f_{2}^\mathrm{S}}{f_{2}^{0}}=\frac{1}{R T}\left( {\mu _{2}^\mathrm{S}} -{\mu _{2}^{0}}\right).\)
It is useful to put the calculation of the solvation free energy using the SMD and SM8 solvation model in the context/language of a conventional molecular simulation free energy calculation. The free energy of solvation is computed as the change in free energy of coupling/decoupling a single solute molecule to solution. When coupling/decoupling a single solute molecule when performing a molecular simulation free energy calculation, the SMD and SM8 calculations assume that the simulation box is approximately the same size when the solute is fully coupled and fully decoupled. This may equivalently be expressed as the change in free energy of taking a single solute molecule from an ideal gas phase (or vacuum) to solution at the same concentration. Additionally, note that in the SMD and SM8 solvation model the solvent is modeled as a continuum; the value of \(N_{1}\) in Eq. 8 is therefore not of importance.
Note that TraPPE-EH does parameterize aniline in ref. [71]. We chose not to use the N LJ parameters from aniline because it is a primary amine, and it was previously shown in ref. [92] that there is an appreciable change in LJ parameters in going from a primary to secondary amine. Also, in ref. [92], the primary amide LJ parameters are the same as the primary amine (i.e., only the charges change). Therefore, we took the secondary amide LJ parameters for N to be the same as for a secondary amine. The H has LJ parameters of 0 in primary and secondary amines and primary amides. The LJ parameters for H in a secondary amide were therefore taken to also be 0 in the present study.
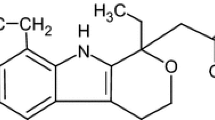
The alkyl groups (CH\(_{3}\), CH\(_{2}\) and CH) were modeled as a single united-atom pseudoatom as a result of the parameterization of the TraPPE-EH force field for n-alkanes which places the LJ site for a hydrogen atom at the center of the corresponding bond [14], and the complication of implementing such a model in a molecular dynamics framework. Note that for the CH group attached to the hydroxyl group in ephedrine, we used CH LJ parameters as in 2-propanol [15]. These differ slightly from the LJ parameters for a CH group for a branched alkane [55]. (The value of \(\sigma\) is smaller).
The exception to this is the value for water which MOSCED doubles (\(v_1=36\) cm\(^3\)/mol) to obtain better agreement with experiment when predicting infinite dilution activity coefficients. We use the value of \(v_1=18\) cm\(^3\)/mol when computing reference solvent normalized activity coefficients using our SMD and SM8 solvation free energies.
References
GROMACS: Fast, flexible, free. http://www.gromacs.org/. Accessed 1 Aug 2013
Minnesota solvation models and solvation software. http://comp.chem.umn.edu/solvation/. Accessed 9 Aug 2015
PyMBAR: Python implementation of the multistate Bennett acceptance Rrtio (MBAR). https://github.com/choderalab/pymbar. Accessed 1 May 2014
Abildskov J (ed) (2005) Solubility and related properties of large complex chemicals part 2: organic solutes ranging from C2 to C41. DECHEMA, Frankfurt am Main
Argones JL, Sanz E, Vega C (2012) Solubility of NaCl in water by molecular simulation revisited. J Chem Phys 136(244):508
Bayly CI, Cieplak P, Cornell WD, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 97:10269–10280
Bennett CH (1976) Efficient estimation of free energy differences from Monte Carlo data. J Comp Phys 22:245–268
Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF (1994) Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem Phys Lett 222:529–539
Blanks RF, Prausnits JM (1964) Thermodynamics of polymer solubility in polar and nonpolar systems. Ind Eng Chem Fundam 3:1–8
Case DA, Cheatham T, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods R (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688
Case DA, Darden TA, Cheatham, III TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Götz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman P Amber 12
Chatterjee K, Dollimore D, Alexander KS (2002) Calculation of vapor pressure curves for hydroxy benzoic acid derivatives using thermogravimetry. Thermochim Acta 392–393:107–117
Chen B, Siepmann JI (1999) Transferable potentials for phase equilibria. 3. Explicit-Hydrogen description of normal alkanes. J Phys Chem B 103:5370–5379
Chen B, Siepmann JI (2006) Microscopic structure and solvation in dry and wet octanol. J Phys Chem B 110:3555–3563
Chen B, Potoff JJ, Siepmann JI (2001) Monte Carlo calculations for alcohols and their mixtures with alkanes. Transferable potentials for phase equilibria. 5. United-atom description of primary, secondary, and tertiary alcohols. J Phys Chem B 105:3093–3104
Chen C, Crafts PA (2006) Correlation and prediction of drug molecule solubility in mixed solvent systems with the nonrandom two-liquid segment activity coefficient (NRTL-SAC) model. Ind Eng Chem Res 45:4816–4824
Chernick MR (2008) Bootstrap methods: a guide for practitioners and researchers. Wiley, Hoboken
Chipot C, Pohorille A (eds) (2007) Free energy calculations: theory and applications in chemistry and biology, Springer series in chemical physics, vol 86. Springer, New York
Chodera JD, Swope WC, Pitera JW, Seok C, Dill KA (2007) Use of the weighted histogram analysis method for the analysis of simulated and parallel tempering simulations. J Chem Theory Comput 3:26–41
Cieplak P, Cornell WD, Bayly C, Kollman PA (1995) Application of the multimolecule and multiconformational RESP methodology to biopolymers: charge derivation for DNA, RNA, and proteins. J Comput Chem 16:1357–1377
Constable DJC, Jimenez-Gonzalez C, Hendersen RK (2007) Perspective on solvent use in the pharmaceutical industry. Org Process Res Dev 11:133–137
Cramer CJ (2002) Essentials of computational chemistry. Wiley, Chichester
Diaz-Rodriguez S, Bozada SM, Phifer JR, Paluch AS (2016) Predicting cyclohexane/water distribution coefficients for the SAMPL5 challenge using MOSCED and the SMD solvation model. J Comput Aided Mol Des. doi:10.1007/s10822-016-9945-9
Draucker LC, Janakat M, Lazzaroni MJ, Bush D, Eckert CA, Frank TC, Olson JD (2007) Experimental determination and model prediction of solid solubility of multifunctional compounds in pure and mixed nonelectrolyte solvents. Ind Eng Chem Res 46:2198–2204
Eaton JW, Bateman D, Hauberg S (2009) GNU Octave version 3.0.1 manual: a high-level interactive language for numerical computations. CreateSpace independent publishing platform. http://www.gnu.org/software/octave/doc/interpreter. ISBN 1441413006
Efron B (1982) The Jackknife, the bootstrap and other resampling plans. Society for Industrial and Applied Mathematics, Philadelphia
Eike DM, Maginn EJ (2006) Atomistic simulation of solid-liquid coexistence for molecular systems: application to triazole and benzene. J Chem Phys 124(164):503
Ferrario M, Ciccotti G, Spohr E, Cartailler T, Turq P (2002) Solubility of KF in water by molecular dynamics using the Kirkwood integration method. J Chem Phys 117:4947–4953
Frank TC, Anderson JJ, Olson JD, Eckert CA (2007) Application of MOSCED and UNIFAC to screen hydrophobic solvents for extraction of hydrogen-bonding organics from aqueous solution. Ind Eng Chem Res 46:4621–4625
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ Gaussian 09, Revision C.01
Fuerst GB, Ley RT, Paluch AS (2015) Calculating the fugacity of pure, low volatile liquids via molecular simulation with application to acetanilide, acetaminophen, and phenacetin. Ind Eng Chem Res 54:9027–9037
Gmehling J (2009) Present status and potential of group contribution methods for process development. J Chem Thermodyn 41:731–747
Gracin S, Brinck T, Rasmuson AC (2002) Prediction of solubility of solid organic compounds in solvents by UNIFAC. Ind Eng Chem Res 41:5114–5124
Grodowska K, Parczewski A (2010) Organic solvents in the pharmaceutical industry. Acta Pol Pharm 67:3–12
Hansen CM (1969) The universality of the solubility parameter. Ind Eng Chem Prod Res Dev 8:2–11
Hess B, Kutzner C, van der Spoel D, Lindal E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447
Hildebrand JH, Prausnitz JM, Scott RL (1970) Regular and related solutions. Van Nostrand Reinhold Company, New York
Hojjati H, Rohani S (2006) Measurement and prediction of solubility of paracetamol in water-isopropanol solution. Part 2. Prediction. Org Process Res Dev 10:1110–1118
Hukkerikar AS, Sarup B, Kate AT, Abildskov J, Sin G, Gani R (2012) Group-contribution+ (gc+) based estimation of properties of pure components: improved property estimation and uncertainty analysis. Fluid Phase Equilib 321:25–43
Jayaraman S, Maginn EJ (2007) Computing the melting point and thermodynamic stability of the orthorhombic and monoclinic crystalline polymorphs of the ionic liquid 1-n-butyl-3-methylimidazolium chloride. J Chem Phys 127(214):504
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926
Klimovich PV, Shirts MR, Mobley DL (2015) Guidelines for analysis of free energy calculations. J Comput Aided Mol Des 29:397–411
Kofke DA, Cummings PT (1997) Quantitative comparison and optimization of methods for evaluating the chemical potential by molecular simulation. Mol Phys 92:973–996
Kofke DA, Cummings PT (1998) Precision and accuracy of staged free-energy perturbation methods for computing the chemical potential by molecular simulation. Fluid Phase Equilib 150–151:41–49
Lazzaroni MJ, Bush D, Eckert CA, Frank TC, Gupta S, Olson JD (2005) Revision of MOSCED parameters and extension to solid solubility calculations. Ind Eng Chem Res 44:4075–4083
Ley RT, Fuerst GB, Redeker BN, Paluch AS (2016) Developing a predictive form of MOSCED for non-electrolyte solids using molecular simulation: application to acetanilide, acetaminophen and phenacetin. Ind Eng Chem Res 55:5415–5430
Lide DR, Milne GWA (eds) (1994) Handbook of Data on Organic Compounds, 3rd edn. CRC Press Inc, Boca Raton
Liu R (ed) (2008) Water-insoluble drug formulation, 2nd edn. CRC Press, Boca Raton
Lu N, Singh JK, Kofke DA (2003) Appropriate methods to combine forward and reverse free-energy perturbation averages. J Chem Phys 118:2977–2984
Marenich AV, Olson RM, Kelly CP, Cramer CJ, Truhlar DG (2007) Self-Consistent Reaction Field Model for Aqueous and Nonaqueous Solutions Based on Accurate Polarized Partial Charges. J Chem Theory Comput 3:2011–2033
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396
Marenich AV, Cramer CJ, Truhlar DG (2013) Generalized born solvation model SM12. J Chem Theory Comput 9:609–620
Marrero J, Abildskov J (eds) (2003) Solubility and related properties of large complex chemicals Part 1: organic solutes ranging from C4 to C40. DECHEMA, Frankfurt am Main
Martin MG, Siepmann JI (1998) Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes. J Phys Chem B 102:2569–2577
Martin MG, Siepmann JI (1999) Novel configurational-bias Monte Carlo method for branched molecules. Transferable potentials for phase equilibria. 2. United-Atom description of branched alkanes. J Phys Chem B 103:4508–4517
Mester Z, Panagiotopoulos AZ (2015) Mean ionic activity coefficients in aqueous NaCl solutions from molecular dynamics simulations. J Chem Phys 142(044):507
Moucka F, Lisal M, Skvor J, Jirsak J, Nezbeda I, Smith WR (2011) Molecular simulation of aqueous electrolyte solubility. 2. Osmotic ensemble Monte Carlo methodology for free energy and solubility calculations and application to NaCl. J Phys Chem B 115:7849–7861
Nordström FL, Rasmuson AC (2008) Determination of the activity of a molecular solute in saturated solution. J Chem Thermodyn 40:1684–1692
Nordström FL, Rasmuson AC (2009) Prediction of solubility curves and melting properties of organic and pharmaceutical compounds. Eur J Pharm Sci 36:330–344
O’Connell JP, Haile JM (2005) Thermodynamics: fundamentals for applications. Cambridge University Press, New York
O’Connell JP, Gani R, Mathias PM, Maurer G, Olson JD, Crafts PA (2009) Thermodynamic property modeling for chemical process and product engineering: some perspectives. Ind Eng Chem Res 48:4619–4637
Paluch AS, Maginn EJ (2013) Predicting the solubility of solid phenanthrene: a combined molecular simulation and group contribution approach. AIChE J 59:2647–2661
Paluch AS, Jayaraman S, Shah JK, Maginn EJ (2010) A method for computing the solubility limit of solids: application to sodium chloride in water and alcohols. J Chem Phys 133(124):504
Phifer JR, Solomon KJ, Young KL, Paluch AS (2016) Computing MOSCED parameters of nonelectrolyte solids with electronic structure methods in SMD and SM8 continuum solvents. AIChE J. doi:10.1002/aic.15413
Poling BE, Prausnitz JM, O’Connell JP (2001) The properties of gases and liquids, 5th edn. McGraw-Hill, New York
Prausnitz JM, Lichtenthaler RN, de Azevedo EG (1999) Molecular thermodynamics of fluid-phase equilibria, 3rd edn. Prentice-Hall Inc, Upper Saddle River
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29:845–854
Rafferty JL, Sun L, Siepmann JI, Schure MR (2010) Investigation of the driving forces for retention in reversed-phase liquid chromatography: Monte Carlo simulations of solute partitioning between n-hexadecane and various aqueous-organic mixtures. Fluid Phase Equilib 290:25–35
Rai N, Siepmann JI (2007) Transferable potentials for phase equilibria. 9. Explicit hydrogen description of benzene and five-membered and six-membered heterocyclic aromatic compounds. J Phys Chem B 111:10790–10799
Rai N, Siepmann JI (2013) Transferable potentials for phase equilibria. 10. Explicit-hydrogen description of substituted benzenes and polycyclic aromatic compounds. J Phys Chem B 117:273–288
Rai N, Bhatt D, Siepmann JI, Fried LE (2008) Monte Carlo simulations of 1,3,5-triamino-2,4,6-trinitrobenzene (TATB): pressure and temperature effects for the solid phase and vapor-liquid phase equilibria. J Chem Phys 129(194):510
Sanz E, Vega C (2007) Solubility of KF and NaCl in water by molecular simulation. J Chem Phys 126(014):507
Schnieders MJ, Baltrusaitis J, Shi Y, Chattree G, Zheng LQ, Yang W, Ren PY (2012) The structure, thermodynamics, and solubility of organic crystals from simulation with a polarizable force field. J Chem Theory Comput 8:1721–1736
Shao Y, Gan Z, Epifanovsky E, Gilbert ATB, Wormit M, Kussmann J, Lange AW, Behn A, Deng J, Feng X, Ghosh D, Goldey M, Horn PR, Jacobson LD, Kaliman I, Khaliullin RZ, Kús T, Landau A, Liu J, Proynov EI, Rhee YM, Richard RM, Rohrdanz MA, Steele RP, Sundstrom EJ, Woodcock HL III, Zimmerman PM, Zuev D, Albrecht B, Alguire E, Austin B, Beran GJO, Bernard YA, Berquist E, Brandhorst K, Bravaya KB, Brown ST, Casanova D, Chang CM, Chen Y, Chien SH, Closser KD, Crittenden DL, Diedenhofen M, DiStasio RA Jr, Dop H, Dutoi AD, Edgar RG, Fatehi S, Fusti-Molnar L, Ghysels A, Golubeva-Zadorozhnaya A, Gomes J, Hanson-Heine MWD, Harbach PHP, Hauser AW, Hohenstein EG, Holden ZC, Jagau TC, Ji H, Kaduk B, Khistyaev K, Kim J, Kim J, King RA, Klunzinger P, Kosenkov D, Kowalczyk T, Krauter CM, Lao KU, Laurent A, Lawler KV, Levchenko SV, Lin CY, Liu F, Livshits E, Lochan RC, Luenser A, Manohar P, Manzer SF, Mao SP, Mardirossian N, Marenich AV, Maurer SA, Mayhall NJ, Oana CM, Olivares-Amaya R, O’Neill DP, Parkhill JA, Perrine TM, Peverati R, Pieniazek PA, Prociuk A, Rehn DR, Rosta E, Russ NJ, Sergueev N, Sharada SM, Sharmaa S, Small DW, Sodt A, Stein T, Stück D, Su YC, Thom AJW, Tsuchimochi T, Vogt L, Vydrov O, Wang T, Watson MA, Wenzel J, White A, Williams CF, Vanovschi V, Yeganeh S, Yost SR, You ZQ, Zhang IY, Zhang X, Zhou Y, Brooks BR, Chan GKL, Chipman DM, Cramer CJ, Goddard WA III, Gordon MS, Hehre WJ, Klamt A, Schaefer HF III, Schmidt MW, Sherrill CD, Truhlar DG, Warshel A, Xua X, Aspuru-Guzik A, Baer R, Bell AT, Besley NA, Chai JD, Dreuw A, Dunietz BD, Furlani TR, Gwaltney SR, Hsu CP, Jung Y, Kong J, Lambrecht DS, Liang W, Ochsenfeld C, Rassolov VA, Slipchenko LV, Subotnik JE, Van Voorhis T, Herbert JM, Krylov AI, Gill PMW, Head-Gordon M (2015) Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol Phys 113:184–215
Shing KS, Chung ST (1987) Computer simulation methods for the calculation of solubility in supercritical extraction systems. J Phys Chem 91:1674–1681
Shirts MR, Chodera JD (2008) Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys 129(124):105
Shirts MR, Pande VS (2005) Solvation free energies of amino acid side chain analogs for common molecular mechanics water models. J Chem Phys 122(134):508
Shirts MR, Bair E, Hooker G, Pande VS (2003) Equilibrium free energies from nonequilibrium measurements using maximum-likelihood methods. Phys Rev Lett 91(140):601
Shirts MR, Pitera JW, Swope WC, Pande VS (2003) Extremely precise free energy calculations of amino acid side chain analogs: comparison of common molecular mechanics force fields for proteins. J Chem Phys 119:5740–5761
Smith JM, Van Ness HC, Abbott MM (2005) Introduction to chemical engineering thermodynamics, 7th edn. McGraw-Hill, New York
Sousa da Silva AW, Vranken WF acpype: AnteChamber PYthon Parser interfacE. http://www.gromacs.org/. Accessed 1 May 2014
Sousa da Silva AW, Vranken WF (2012) ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res Notes 5:367
Steinbrecher T, Mobley DL, Case DA (2007) Nonlinear scaling schemes for Lennard-Jones interactions in free energy calculations. J Chem Phys 127(214):108
Stephenson RM, Malanowski S (1987) Handbook of the thermodynamics of organic compounds. Elsevier, New York
Storn R, Price K (1997) Differential evolution—simple and efficient heuristic for global optimization over continuous spaces. J Global Optim 11:341–359
Stubbs JM, Potoff JJ, Siepman JI (2004) Transferable potentials for phase qquilibria. 6. United-atom description for ethers, glycols, ketones, and aldehydes. Phys Chem B 108:17596–17605
Thomas ER, Eckert CA (1984) Prediction of limiting activity coefficients by a modified separation of cohesive energy density model and UNIFAC. Ind Eng Chem Proc Des Dev 23:194–209
Walas SM (1985) Phase equilibria in chemical engineering. Butterworth Publishers, Stoneham
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174
Wang J, Wang W, Kollman PA, Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graphics Modell 25:247–260
Wick CD, Martin MG, Siepmann JI (2000) Transferable potentials for phase equilibria. 4. United-atom description of linear and branched alkenes and alkylbenzenes. J Phys Chem B 104:8008–8016
Wick CD, Stubbs JM, Rai N, Siepmann JI (2005) Transferable potentials for phase equilibria. 7. Primary, secondary, and tertiary amines, nitroalkanes and nitrobenzene, nitriles, amides, pyridine, and pyrimidine. J Phys Chem B 109:18974–18982
Winget P, Hawkins GD, Cramer CJ, Truhlar DG (2000) Prediction of vapor pressures from self-solvation free energies calculated by the sm5 series of universal solvation models. J Phys Chem B 104:4726–4734
Yang H, Thati J, Rasmuson AC (2012) Thermodynamics of molecular solids in organic solvents. J Chem Thermodyn 48:150–159
Zhao Y, Truhlar DG (2008) The M06 theory of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account 120:215–241
Acknowledgements
C.E.C., J.R.P, and E.J.O. are thankful for financial support through the Undergraduate Summer Scholars (USS) program through the Office of Research for Undergraduates at Miami University. B.T.R., C.E.C. and R.T.L. gratefully acknowledge financial support from the Miami University College of Engineering and Computing. G.G.N., L.F.S. and A.K.P.B. gratefully acknowledge financial support through the Brazil Scientific Mobility Program, sponsored by CAPES and CNPq. Computing support was provided by the Ohio Supercomputer Center and Miami University’s Research Computing Support group.
Author information
Authors and Affiliations
Corresponding author
Additional information
Courtney E. Cox, Jeremy R. Phifer, Larissa Ferreira da Silva, Gabriel Gonçalves Nogueira, Ryan T. Ley, Elizabeth J. O’Loughlin, Ana Karolyne Pereira Barbosa, Brett T. Rygelski contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cox, C.E., Phifer, J.R., Ferreira da Silva, L. et al. Combining MOSCED with molecular simulation free energy calculations or electronic structure calculations to develop an efficient tool for solvent formulation and selection. J Comput Aided Mol Des 31, 183–199 (2017). https://doi.org/10.1007/s10822-016-0001-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-016-0001-6