
Abstract
In small molecule drug discovery projects, the receptor structure is not always available. In such cases it is enormously useful to be able to align known ligands in the way they bind in the receptor. Here we shall present an algorithm for the alignment of multiple small molecule ligands. This algorithm takes pre-generated conformers as input, and proposes aligned assemblies of the ligands. The algorithm consists of two stages: the first stage is to perform alignments for each pair of ligands, the second stage makes use of the results from the first stage to build up multiple ligand alignment assemblies using a novel iterative procedure. The scoring functions are improved versions of the one mentioned in our previous work. We have compared our results with some recent publications. While an exact comparison is impossible, it is clear that our algorithm is fast and produces very competitive results.








Similar content being viewed by others
References
Cramer RD, Patterson DE, Bunce JD (1988) Comparative molecular field analysis (CoMFA). 1. effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 110:5959–5967
Lemmen C, Langauer T (2000) Computational methods for the structural alignment of molecules. J Comput Aided Mol Des 14:215–232
Wolber G, Seidel T, Bendix F, Langer T (2008) Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov Today 13:23–39
Leach AR, Gillet VJ, Lewis RA, Taylor R (2010) Three-dimensional pharmacophore methods in drug discovery. J Med Chem 53:539–558
Jones G, Willett P, Glen RC (1995) A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J Comput Aided Mol Des 9:532–549
Barnum D, Greene J, Smellie A, Sprague P (1996) Identification of common functional configurations among molecules. J Chem Inf Comput Sci 36:563–571
Lemmen C, Lengauer T, Klebe G (1998) FLEXS: a method for fast flexible ligand superposition. J Med Chem 41:4502–4520
Labute P, Williams C, Feher M, Sourial E, Schmidt JM (2001) Flexible alignment of small molecules. J Med Chem 44:1483–1490
ROCS, from OpenEye Scientific Software, http://www.eyesopen.com
Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA (2006) PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des 20:647–671
Gardiner EJ, Cosgrove DA, Taylor R, Gillet VJ (2009) Multiobjective optimization of pharmacophore hypotheses: bias toward low-energy conformations. J Chem Inf Model 49:2761–2773
Jones G (2010) GAPE: an improved genetic algorithm for pharmacophore elucidation. J Chem Inf Model 50:2001–2018
Taylor R, Cole JC, Cosgrove DA, Gardiner EJ, Gillet VJ, Korb O (2012) Development and validation of an improved algorithm for overlaying flexible molecules. J Comput Aided Mol Des 26:451–472
Korb O, Monecke P, Hessler G, Stützle T, Exner TE (2010) pharmACOphore: multiple flexible ligand alignment based on ant colony optimization. J Chem Inf Model 50:1669–1681
Moser D, Wittmann SK, Kramer J, Blöcher R, Achenbach J, Pogoryelov D, Proschak E (2015) PENG: a neural gas-based approach for pharmacophore elucidation. Method design, validation, and virtual screening for novel ligands of LTA4H. J Chem Inf Model 55:284–293
Shin W, Hyun SA, Chae CH, Chon JK (2009) Flexible alignment of small molecules using the penalty method. J Chem Inf Model 49:1879–1888
Tosco P, Balle T, Shiri F (2011) Open3DALIGN: an open-source software aimed at unsupervised ligand alignment. J Comput Aided Mol Des 25:777–783
Diller DJ, Connell ND, Welsh WJ (2015) Avalanche for shape and feature-based virtual screening with 3D alignment. J Comput Aided Mol Des 29:1015–1024
Kawabata T, Nakamura H (2014) 3D flexible alignment using 2D maximum common substructure: dependence of prediction accuracy on target-reference chemical similarity. J Chem Inf Model 54:1850–1863
Vainio MJ, Puranen JS, Johnson MS (2009) ShaEP: molecular overlay based on shape and electrostatic potential. J Chem Inf Model 49:492–502
Cross S, Baroni M, Goracci L, Cruciani G (2012) GRID-based three-dimensional pharmacophores I: FLAPpharm, a novel approach for pharmacophore elucidation. J Chem Inf Model 52:2587–2598
Leherte L, Vercauteren DP (2012) Smoothed Gaussian molecular fields: an evaluation of molecular alignment problems. Theor Chem Acc 131:1259–1275
Sastry GM, Dixon SL, Sherman W (2011) Rapid shape-based ligand alignment and virtual screening method based on atom/feature-pair similarities and volume overlap scoring. J Chem Inf Model 51:2455–2466
Jones G, Gao Y, Sage CR (2009) Elucidating molecular overlays from pairwise alignments using a genetic algorithm. J Chem Inf Model 49:1847–1855
Klabunde T, Giegerich C, Evers A (2012) MARS: computing three-dimensional alignments for multiple ligands using pairwise similarities. J Chem Inf Model 52:2022–2030
Chan SL, Labute P (2010) Training a scoring function for the alignment of small molecules. J Chem Inf Model 50:1724–1735
Bron C, Kerbosch J (1973) Algorithm 457—finding all cliques of an undirected graph. Commun ACM 16:575–577
Kabsch W (1978) A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallogr A 34:827–828
Cross S, Ortuso F, Baroni M, Costa G, Distinto S, Moraca F, Alcaro S, Cruciani G (2012) GRID-based three-dimensional pharmacophores II: pharmbench, a benchmark data set for evaluating pharmacophore elucidation methods. J Chem Inf Model 52:2599–2608
Giangreco I, Cosgrove DA, Packer MJ (2013) An extensive and diverse set of molecular overlays for the validation of pharmacophore programs. J Chem Inf Model 53:852–866
The Protein Data Bank. http://www.rcsb.org
Giangreco I, Olsson TSG, Cole JC, Packer MJ (2014) Assessment of a cambridge structural database-driven overlay program. J Chem Inf Model 54:3091–3098
The RDKit. http://www.rdkit.org, by Greg Landrum et al.
Vainio MJ, Johnson MS (2007) Generating conformer ensembles using a multiobjective genetic algorithm. J Chem Inf Model 47:2462–2474
Schärfer C, Schulz-Gasch T, Hert J, Heinzerling L, Schulz B, Inhester T, Stahl M, Rarey M (2013) CONFECT: conformations from an expert collection of torsion patterns. ChemMedChem 8:1690–1700
Richmond NJ, Abrams CA, Wolohan PR, Abrahamian E, Willett P, Clark RD (2006) GALAHAD: 1. pharmacophore identification by hypermolecular alignment of ligands in 3D. J Comput Aided Mol Des 20:567–587
Acknowledgements
The author would like to thank Dr. Christian Lemmen and the authors of Confect [35], and Dr. Mikko Vainio and other authors of Balloon [34]. This work would have been impossible without free access to these two conformer generators. Other indispensible freeware include the RDKit [33], the Yasara viewer (http://www.yasara.org), and the JMol Viewer (http://jmol.sourceforge.net). The author also thanks Drs. Alex Clark and Bill Myrtle for help with proof-reading.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix
Appendix
Termination criterion for simultaneous multiple pose generation
After each cycle (each query triplet), the pair pose registers may be updated. The raw score improvement is defined to be the average (over all registers) of the improvement of the score at the fifth slot (i.e., the fifth best score in the register) over this cycle. The first slot is not used because it does not get updated very often (for the first slot to be updated, a new pose with a score better than any of the existing ones must be found). Position 5 is used because each register is guaranteed to have more than five slots, as per Eq. (2). In principle, we would terminate the iteration when the score improvement, SI, becomes too small. We noticed that some cycles produced very small SIs although there was still significant room for improvement. In order to smooth out the fluctuation, we average out the SI of the current cycle with the SIs of a few past cycles to create an effective SI, and use this to determine termination. Let \({n_{curr}}\) be the number of compatible triplets (which equals the number of conformer poses in the grand assembly) encountered in the current cycle. Let \({n_{avg}}\) be the hitherto average number of compatible triplets per cycle (i.e., \({n_{avg}}\) is the sum of all \({n_{curr}}\)′s encountered so far, divided by the number of cycles). The effective SI of the current cycle, \({s_{curr}}\), is given by
where \({s_{prev}}\) is the effective SI of the previous cycle, and \({s_0}\) is the raw score improvement, as described above. In other words, \({s_{curr}}\) is a weighted average of \({s_{prev}}\) and \({s_0}\). \({s_0}\) is weighted less if there are few compatible triplets in the current cycle. This should make the effective SI, \({s_{curr}}\), less prone to statistical fluctuations than the raw score improvement, \(~{s_0}\).
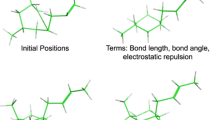
Iteration terminates when the effective SI drops below a cut-off value, \({s_{cut}}\). \({s_{cut}}\) is set to a value of 0.01. To put this in perspective, the pairwise score (according to any scoring scheme) of one hydrophobic atom overlapping on another is 2.0.
For the first cycle, the value of \({s_{prev}}\) in Eq. (3) is set to \({2^5} \times {s_{cut}}\). 5 is the index of the slot used for determining the raw score improvement, \({s_0}\), as mentioned above. This way, if \({s_0}\) is 0 and \({n_{curr}}\) remains unchanged over the cycles (implying that \({n_{avg}}\) always equals \({n_{curr}}\)), the iteration would go on for 5 cycles before terminating. Put in another way, if only one slot of each pair pose register is filled per iteration cycle, there is time for the register to be filled to the fifth slot, where \({s_0}\) is determined.
Choosing random query triplets for simultaneous multiple pose generation
Early in our research, we simply chose a random triplet from a random conformer of a random ligand. We found this inefficient. Since we would like each pair pose register to be at least partially filled, we can bias the choice of the ligands to encourage ligands with empty registers to be chosen. First, ligands having fewer than three representative points are not involved, because they do not have representative triplets. An array is constructed out of the indices of all ligands having at least three representative points. Each index appears once. We then go through all ligand pairs. For each ligand pair whose pair pose register is still empty, three copies of the index of each ligand in the pair are inserted into the array. (Experiments showed that using five instead of three has little effect on the results.) Finally, a random element from this array is selected to give a random ligand. Then a random conformer of this ligand is chosen, and a random triplet of representative points of this conformer is chosen. Similar to the systematic pose generation method, triplets that are geometrically too small are never chosen. The diameter (see “Systematic pose generation” section) of a chosen triplet has to be at least half that of the smallest diameter amongst the representative point sets of all conformers of all ligands.
Starting points for multiple ligand alignment
As described in the section “Generating multiple ligand assemblies”, one base template conformer gives rise to one starting point assembly, and subsequently one solution assembly. We discovered that using a maximum of 250 starting points strikes a balance between good results and speed. Before the iteration begins, each conformer of each ligand is used as a base template to generate a starting assembly. Given one base template conformer, for each of the other ligands, the best scoring pairwise alignment pose between the ligand and the base template is used to build the starting assembly. It is possible that no pairwise alignment pose exists in the pair pose register between the base template conformer and the ligand. If this is the case, the missing ligand count of this base template will be increased by one. After all the non-missing ligands have been positioned in the assembly, each missing ligand will be positioned using the non-missing ligands as helpers, one at a time. For each missing ligand, amongst all poses generated using the helpers, the one resulting in the best score for the assembly (all non-missing ligands plus the current ligand) is used. Any ligands still missing after this step will be positioned randomly. The multiple ligand alignment score of the starting point assembly will then be calculated, using fast scoring. The full list of starting point assemblies will then be sorted, first by the missing ligand count, then by the multiple ligand alignment score. If there are more than 250 assemblies, the 250 with the smallest missing ligand counts and the best scores are used as starting points. In a real run, there is of course no need to patch the missing ligands if there are already enough starting points without any missing ligands.
Rights and permissions
About this article

Cite this article
Chan, S.L. MolAlign: an algorithm for aligning multiple small molecules. J Comput Aided Mol Des 31, 523–546 (2017). https://doi.org/10.1007/s10822-017-0023-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-017-0023-8