
Abstract
Recent advances in the development of low-cost quantum chemical methods have made the prediction of conformational preferences and physicochemical properties of medium-sized drug-like molecules routinely feasible, with significant potential to advance drug discovery. In the context of the SAMPL6 challenge, macroscopic pKa values were blindly predicted for a set of 24 of such molecules. In this paper we present two similar quantum chemical based approaches based on the high accuracy calculation of standard reaction free energies and the subsequent determination of those pKa values via a linear free energy relationship. Both approaches use extensive conformational sampling and apply hybrid and double-hybrid density functional theory with continuum solvation to calculate free energies. The blindly calculated macroscopic pKa values were in excellent agreement with the experiment.







Similar content being viewed by others
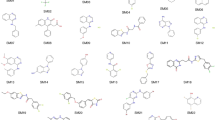
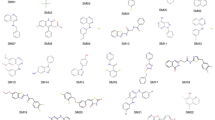
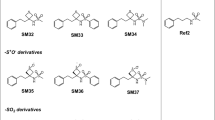
References
Manallack DT (2007) The pKa distribution of drugs: application to drug discovery. Perspect Med Chem 1:1177391X0700100003
D3R. Sampl6 homepage. https://drugdesigndata.org/about/sampl6. Accessed 30 May 2018
Nicholls A, Mobley DL, Guthrie JP, Chodera JD, Bayly CI, Cooper MD, Pande VS (2008) Predicting small-molecule solvation free energies: an informal blind test for computational chemistry. J Med Chem 51(4):769–779
Mobley DL, Bayly CI, Cooper MD, Dill KA (2009) Predictions of hydration free energies from all-atom molecular dynamics simulations. J Phys Chem B 113(14):4533–4537
Klimovich PV, Mobley DL (2010) Predicting hydration free energies using all-atom molecular dynamics simulations and multiple starting conformations. J Comput-Aided Mol Des 24(4):307–316
Geballe MT, Skillman AG, Nicholls A, Guthrie JP, Taylor Peter J (2010) The SAMPL2 blind prediction challenge: introduction and overview. J Comput-Aided Mol Des 24(4):259–279
Geballe MT, Guthrie JP (2012) The SAMPL3 blind prediction challenge: transfer energy overview. J Comput-Aided Mol Des 26(5):489–496
Mobley DL, Wymer KL, Lim NM, Guthrie JP (2014) Blind prediction of solvation free energies from the SAMPL4 challenge. J Comput-Aided Mol Des 28(3):135–150
Bannan CC, Burley KH, Chiu M, Shirts MR, Gilson MK, Mobley DL (2016) Blind prediction of cyclohexane-water distribution coefficients from the SAMPL5 challenge. J Comput-Aided Mol Des 30(11):927–944
Darvey IG (1995) The assignment of pKa values to functional groups in amino acids. Biochem Educ 23(2):80–82
Bodner GM (1986) Assigning the pKa’s of polyprotic acids. J Chem Educ 63(3):246
Murray R (1995) Microscopic equilibria. Anal Chem 67(15):462a
Grimme S (2012) Supramolecular binding thermodynamics by dispersioncorrected density functional theory. Chem Eur J 18(32):9955–9964
Pliego JR (2003) Thermodynamic cycles and the calculation of pKa. Chem Phys Lett 367(1):145–149
da Silva CO, da Silva EC, Nascimento MAC (1999) Ab initio calculations of absolute pKa values in aqueous solution i. Carboxylic acids. J Phys Chem A 103(50):11194–11199
Dissanayake DP, Senthilnithy R (2009) Thermodynamic cycle for the calculation of ab initio pKa values for hydroxamic acids. J Mol Struct THEOCHEM 910(1):93–98
Klamt A, Eckert F, Diedenhofen M, Beck ME (2003) First principles calculations of aqueous pKa values for organic and inorganic acids using COSMO-RS reveal an inconsistency in the slope of the pKa scale. J Phys Chem A 107(44):9380–9386
Grimme S, Bannwarth C, Shushkov P (2017) A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (z = 186). J Chem Theory Comput 13(5):1989–2009
Ripin DH, Evans DA Evans pKa table. http://evans.rc.fas.harvard.edu/pdf/evans_pKa_table.pdf. Accessed 30 May 2018
Grimme S, Bannwarth C, Dohm S, Hansen A, Pisarek J, Pracht P, Seibert Jakob, Neese F (2017) Fully automated quantum-chemistry-based computation of spin-spin coupled nuclear magnetic resonance spectra. Angew Chem Int Ed 56(46):14763−14769
Pracht P, Bauer CA, Grimme S (2017) Automated and efficient quantum chemical determination and energetic ranking of molecular protonation sites. J Comput Chem 38(30):2618–2631
Grimme S, Brandenburg JG, Bannwarth C, Hansen A (2015) Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J Chem Phys 143(5):054107
Klamt A, Diedenhofen M (2015) Calculation of solvation free energies with DCOSMO-RS. J Phys Chem A 119(21):5439–5445
Eckert F, Klamt A (2004) Fast solvent screening via quantum chemistry: COSMO-RS approach. AIChE J 48(2):369–385
Klamt A (2018) The COSMO and COSMO-RS solvation models. Wiley Interdiscip Rev-Comput Mol Sci 8(1):e1338
Kozuch S, Gruzman D, Martin JML (2010) DSD-BLYP: a general purpose double hybrid density functional including spin component scaling and dispersion correction. J Phys Chem C 114(48):20801–20808
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132(15):154104
Grimme S, Ehrlich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32(7):1456–1465
Rappoport D, Furche F (2010) Property-optimized gaussian basis sets for molecular response calculations. J Chem Phys 133(13):134105
Furche F, Ahlrichs R, Hättig C, Klopper W, Sierka M, Weigend F (2014) Turbomole. Wiley Interdiscip Rev-Comput Mol Sci 4(2):91–100
Neese F (2012) The ORCA program system. Wiley Interdiscip Rev-Comput Mol Sci 2(1):73–78
Udvarhelyi A, Wilcken R Manuscript in preparation
Molecular Networks GmbH, Nuremberg, Germany. Corina v3.6. https://www.mn-an.com/. Accessed 30 May 2018
Schrödinger, LLC, New York, NY (2017) Schrödinger release 2017-3: MacroModel
Chang G, Guida WC, Still WC (1989) An internal-coordinate monte carlo method for searching conformational space. J Am Chem Soc 111(12):4379–4386
Saunders M, Houk KN, Wu YD, Still WC, Lipton Mark, Chang G, Guida WC (1990) Conformations of cycloheptadecane. A comparison of methods for conformational searching. J Am Chem Soc 112(4):1419–1427
Jorgensen WL, Maxwell DS, Tirado-Rives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118(45):11225–11236
Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 105(28):6474–6487
Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM (2005) Integrated modeling program, applied chemical theory (IMPACT). J Comput Chem 26(16):1752–1780
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33:8822–8824
Schäfer A, Horn H, Ahlrichs R (1992) Fully optimized contracted gaussian basis sets for atoms Li to Kr. J Chem Phys 97(4):2571–2577
Klamt A, Schüürmann G (1993) COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans 2:799–805
Zhao Y, Truhlar DG (2005) Design of density functionals that are broadly accurate for thermochemistry, thermochemical kinetics, and nonbonded interactions. J Phys Chem A 109(25):5656–5667
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77:3865–3868
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305
Bochevarov AD, Watson MA, Greenwood JR, Philipp DM (2016) Multiconformation, density functional theory-based pKa prediction in application to large, flexible organic molecules with diverse functional groups. J Chem Theory Comput 12(12):6001–6019
Eckert F, Klamt A (2006) Accurate prediction of basicity in aqueous solution with COSMO-RS. J Comput Chem 27(1):11–19
Klicic JJ, Friesner RA, Liu SY, Guida WC (2002) Accurate prediction of acidity constants in aqueous solution via density functional theory and self-consistent reaction field methods. J Phys Chem A 106(7):1327–1335
Andersson MP, Jensen JH, Svane SSL (2013) Predicting pKa for proteins using COSMO-RS. PeerJ 1:e198
Kromann JC, Larsen F, Moustafa H, Jensen JH (2016) Prediction of pKa values using the PM6 semiempirical method. PeerJ 4:e2335
Muckerman JT, Skone JH, Ning M, Wasada-Tsutsui Y (2013) Toward the accurate calculation of pKa values in water and acetonitrile. Biochim Biophys Acta Bioenerg 1827(8):882–891
Namazian M, Heidary H (2003) Ab initio calculations of pKa values of some organic acids in aqueous solution. J Mol Struct THEOCHEM 620(2):257–263
Goerigk L, Hansen A, Bauer C, Ehrlich S, Najibi A, Grimme S (2017) A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys Chem Chem Phys 19:32184–32215
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396
Marenich AV, Cramer CJ, Truhlar DG (2009) Performance of SM6, SM8, and SMD on the SAMPL1 test set for the prediction of small-molecule solvation free energies. J Phys Chem B 113(14):4538–4543
Ribeiro RF, Marenich AV, Cramer CJ, Truhlar DG (2010) Prediction of SAMPL2 aqueous solvation free energies and tautomeric ratios using the SM8, SM8AD, and SMD solvation models. J Comput-Aided Mol Des 24(4):317–333
Molecular Discovery Ltd., Borehamwood, UK. Moka v2.5.4. https://www.mn-an.com/. Accessed 30 May 2018
D3R (2018) SAMPL6 NMR characterization https://github.com/MobleyLab/SAMPL6/tree/master/physical_pro-perties/pKa/experimental_data/NMR_microstate_determination. Accessed 30 May 2018
Adamo C, Barone V (1999) Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys 110(13):6158–6170
Ernzerhof M, Scuseria GE (1999) Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J Chem Phys 110(11):5029–5036
Milletti F, Storchi L, Sforna G, Cruciani G (2007) New and original pKa prediction method using grid molecular interaction fields. J Chem Inf Model 47(6):2172–2181
Cruciani G, Milletti F, Storchi L, Sforna G, Goracci Laura (2009) In silico pKa prediction and ADME profiling. Chem Biodivers 6(11):1812–1821
Gedeck P, Yipin L, Skolnik S, Rodde S, Dollinger G, Jia W, Berellini G, Vianello R, Faller B, Lombardo F (2015) Benefit of retraining pKa models studied using internally measured data. J Chem Inf Model 55(7):1449–1459
Bannwarth C, Ehlert S, Grimme S Manuscript in preparation
Caldeweyher E, Bannwarth C, Grimme S (2017) Extension of the D3 dispersion coefficient model. J Chem Phys 147(3):034112
Acknowledgements
We thank Wolfgang Zipfel and the NIBR NX Scientific Computing team for generous allocation of and help with HPC resources. We thank Jens Reinisch, Uwe Huniar, Michael Diedenhofen and the whole COSMOlogic team for helpful discussions. This work was supported by the German Research Foundation (DFG) through a Leibniz prize to S.G.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Pracht, P., Wilcken, R., Udvarhelyi, A. et al. High accuracy quantum-chemistry-based calculation and blind prediction of macroscopic pKa values in the context of the SAMPL6 challenge. J Comput Aided Mol Des 32, 1139–1149 (2018). https://doi.org/10.1007/s10822-018-0145-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-018-0145-7