
Abstract
Prediction of drug-protein binding is critical for virtual drug screening. Many deep learning methods have been proposed to predict the drug-protein binding based on protein sequences and drug representation sequences. However, most existing methods extract features from protein and drug sequences separately. As a result, they can not learn the features characterizing the drug-protein interactions. In addition, the existing methods encode the protein (drug) sequence usually based on the assumption that each amino acid (atom) has the same contribution to the binding, ignoring different impacts of different amino acids (atoms) on the binding. However, the event of drug-protein binding usually occurs between conserved residue fragments in the protein sequence and atom fragments of the drug molecule. Therefore, a more comprehensive encoding strategy is required to extract information from the conserved fragments.
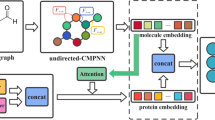
In this paper, we propose a novel model, named FragDPI, to predict the drug-protein binding affinity. Unlike other methods, we encode the sequences based on the conserved fragments and encode the protein and drug into a unified vector. Moreover, we adopt a novel two-step training strategy to train FragDPI. The pre-training step is to learn the interactions between different fragments using unsupervised learning. The fine-tuning step is for predicting the binding affinities using supervised learning. The experiment results have illustrated the superiority of FragDPI.
Similar content being viewed by others


Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.References
Swinney D C, Anthony J. How were new medicines discovered? Nature Reviews Drug Discovery, 2011, 10(7): 507–519
Gupta S, Jadaun A, Kumar H, Raj U, Varadwaj P K, Rao A R. Exploration of new drug-like inhibitors for serine/threonine protein phosphatase 5 of Plasmodium falciparum: a docking and simulation study. Journal of Biomolecular Structure and Dynamics, 2015, 33(11): 2421–2441
Yuriev E, Agostino M, Ramsland P A. Challenges and advances in computational docking: 2009 in review. Journal of Molecular Recognition, 2011, 24(2): 149–164
Huang K, Fu T, Glass L M, Zitnik M, Xiao C, Sun J. DeepPurpose: a deep learning library for drug-target interaction prediction. Bioinformatics, 2020, 36(22–23): 5545–5547
Huang K, Xiao C, Glass L M, Sun J. MolTrans: molecular interaction transformer for drug-target interaction prediction. Bioinformatics, 2021, 37(6): 830–836
Zhao Q, Xiao F, Yang M, Li Y, Wang J. AttentionDTA: prediction of drug—target binding affinity using attention model. In: Proceedings of 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2019, 64–69
Liao Z, You R, Huang X, Yao X, Huang T, Zhu S. DeepDock: enhancing ligand-protein interaction prediction by a combination of ligand and structure information. In: Proceedings of 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2019, 311–317
Bai F, Morcos F, Cheng R R, Jiang H, Onuchic J N. Elucidating the druggable interface of protein-protein interactions using fragment docking and coevolutionary analysis. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113(50): E8051–E8058
Yao H, Song Y, Chen Y, Wu N, Xu J, Sun C, Zhang J, Weng T, Zhang Z, Wu Z, Cheng L, Shi D, Lu X, Lei J, Crispin M, Shi Y, Li L, Li S. Molecular architecture of the SARS-CoV-2 virus. Cell, 2020, 183(3): 730–738.e13
Shu X, Royant A, Lin M Z, Aguilera T A, Lev-Ram V, Steinbach P A, Tsien R Y. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science, 2009, 324(5928): 804–807
Pahikkala T, Airola A, Pietila S, Shakyawar S, Szwajda A, Tang J, Aittokallio T. Toward more realistic drug-target interaction predictions. Briefings in Bioinformatics, 2015, 16(2): 325–337
Zheng X, Ding H, Mamitsuka H, Zhu S. Collaborative matrix factorization with multiple similarities for predicting drug-target interactions. In: Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2013, 1025–1033
Özturk H, Özgür A, Ozkirimli E. DeepDTA: deep drug-target binding affinity prediction. Bioinformatics, 2018, 34(17): i821–i829
Nguyen T, Le H, Venkatesh S. GraphDTA: prediction of drug—target binding affinity using graph convolutional networks. BioRxiv, 2019: 684662
Devlin J, Chang M W, Lee K, Toutanova K. BERT: pre-training of deep bidirectional transformers for language understanding. In: Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies. 2019, 4171–4186
Dong L, Yang N, Wang W, Wei F, Liu X, Wang Y, Gao J, Zhou M, Hon H W. Unified language model pre-training for natural language understanding and generation. In: Proceedings of the 33rd International Conference on Neural Information Processing Systems. 2019, 1170
Radford A, Wu J, Child R, Luan D, Amodei D, Sutskever I. Language models are unsupervised multitask learners. OpenAI blog, 2019, 1(8): 9
Raffel C, Shazeer N, Roberts A, Lee K, Narang S, Matena M, Zhou Y, Li W, Liu P J. Exploring the limits of transfer learning with a unified text-to-text transformer. Journal of Machine Learning Research, 2020, 21: 1–67
Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez A N, Kaiser L, Polosukhin I. Attention is all you need. In: Proceedings of the 31st International Conference on Neural Information Processing Systems. 2017, 6000–6010
Karimi M, Wu D, Wang Z, Shen Y. DeepAffinity: interpretable deep learning of compound—protein affinity through unified recurrent and convolutional neural networks. Bioinformatics, 2019, 35(18): 3329–3338
Liu T, Lin Y, Wen X, Jorissen R N, Gilson M K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Research, 2007, 35(S1): D198–D201
Kuhn M, Von Mering C, Campillos M, Jensen L J, Bork P. STITCH: interaction networks of chemicals and proteins. Nucleic Acids Research, 2008, 36(S1): D684–D688
Suzek B E, Wang Y, Huang H, McGarvey P B, Wu C H, UniProt Consortium. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics, 2015, 31(6): 926–932
Li M, Lu Z, Wu Y, Li Y. BACPI: a bi-directional attention neural network for compound—protein interaction and binding affinity prediction. Bioinformatics, 2022, 38(7): 1995–2002
Leonard T A, Różycki B, Saidi L F, Hummer G, Hurley J H. Crystal structure and allosteric activation of protein kinase C βII. Cell, 2011, 144(1): 55–66
Sutton R B, Sprang S R. Structure of the protein kinase cβ phospholipid-binding C2 domain complexed with Ca2+. Structure, 1998, 6(11): 1395–1405
Thao T T N, Labroussaa F, Ebert N, V’kovski P, Stalder H, Portmann J, Kelly J, Steiner S, Holwerda M, Kratzel A, Gultom M, Schmied K, Laloli L, Hüsser L, Wider M, Pfaender S, Hirt D, Cippà V, Crespo-Pomar S, Schröder S, Muth D, Niemeyer D, Corman V M, Müller M A, Drosten C, Dijkman R, Jores J, Thiel V. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature, 2020, 582(7813): 561–565
Tzenaki N, Papakonstanti E A. p110δ PI3 kinase pathway: emerging roles in cancer. Frontiers in Oncology, 2013, 3: 40
Takahashi Y, Hayakawa A, Sano R, Fukuda H, Harada M, Kubo R, Okawa T, Kominato Y. Histone deacetylase inhibitors suppress ACE2 and ABO simultaneously, suggesting a preventive potential against COVID-19. Scientific Reports, 2021, 11(1): 3379
Volz H P, Gleiter C H. Monoamine oxidase inhibitors. Drugs & Aging, 1998, 13(5): 341–355
Kumar A, Redondo-Muñoz J, Perez-García V, Cortes I, Chagoyen M, Carrera A C. Nuclear but not cytosolic phosphoinositide 3-kinase beta has an essential function in cell survival. Molecular and Cellular Biology, 2011, 31(10): 2122–2133
Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFA0904303).
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhihui Yang is a PhD candidate in the School of Computer Science, Wuhan University, China. His current research interests include synthetic biology, deep learning, metabolic pathway reconstruction, and metabolic flux analysis.
Juan Liu is a professor in the School of Computer Science, Wuhan University, China. Her research interests include machine learning, data mining, bioinformatics, pattern recognition, and artificial intelligence methods for medicine.
Xuekai Zhu is a master’s student in the School of Computer Science, Wuhan University, China. His current research interests are in artificial intelligence methods for bioinformatics.
Feng Yang is a PhD candidate in the School of Computer Science, Wuhan University, China. His current research interests include machine learning, retrosynthesis prediction and metabolic pathway design.
Qiang Zhang is a PhD candidate in the School of Computer Science, Wuhan University, China. Her current research interests include retrosynthesis prediction, metabolic pathway design, bioinformatics, and machine learning.
Hayat Ali Shah received his MS degree in Computer Science from Virtual University of Pakistan, Pakistan in 2018. He is currently a PhD candidate in the School of Computer Science, Wuhan University, China. His research interests are simulated alignments, multiple sequence alignments, machine learning, prediction and reconstruction of metabolic pathways.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Yang, Z., Liu, J., Zhu, X. et al. FragDPI: a novel drug-protein interaction prediction model based on fragment understanding and unified coding. Front. Comput. Sci. 17, 175903 (2023). https://doi.org/10.1007/s11704-022-2163-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11704-022-2163-9