

Abstract
Structure-based methods that employ principles of de novo design can be used to construct small organic molecules from-scratch, using pre-existing fragment libraries to sample chemical space, and are an important class of computational algorithms for drug-lead discovery. Here, we present a powerful new design method for DOCK6 that employs a Descriptor Driven De Novo strategy (termed D3N) in which user-defined cheminformatics descriptors (and their target ranges) are calculated at each layer of growth using the open-source toolkit RDKit. The objective is to tailor ligand growth towards desirable regions of chemical space. The approach was extensively validated through: (1) comparison of cheminformatics descriptors computed using the new DOCK6/RDKit interface versus the standard Python/RDKit installation, (2) examination of descriptor distributions generated using D3N growth under different conditions (target ranges and environments), and (3) construction of ligands with very tight (pinpoint) descriptor ranges using clinically-relevant compounds as a reference. Our testing confirms that the new DOCK6/RDKit integration is robust, showcases how the new D3N routines can be used to direct sampling around user-defined chemical spaces, and highlights the utility of on-the-fly descriptor calculations for ligand design to important drug targets.
Graphical Abstract
Consideration of cheminformatics descriptors when developing and refining small molecule candidates is a critical component of modern drug discovery. In this study, a new interface for the program DOCK6 is presented which enables descriptors computed using the open-source package RDKit to bias de novo design of new ligands during the layer-by-layer growth process. The interface can be used to enrich ensembles towards a specific range for a single property or multiple properties simultaneously.
Introduction
Virtual screening methods are a commonly used and well-validated class of computational tools to help screen libraries of pre-existing purchasable (or likely synthesizable) small organic molecules to a drug target (usually a protein) prior to experimental testing.1 Virtual screening protocols typically involve generation and evaluation of hundreds to thousands of conformations (poses) for every ligand that is docked to the target and identifies those that are most compatible according to physical interactions within the binding site and/or other user-specified criteria. Despite its effectiveness, exhaustive docking of large libraries such as ZINC,2–4 which continue to grow at an astounding rate, can be prohibitive for many users who lack the infrastructures to screen libraries on the order of 107 to 109 molecules. Further, given that the chemical space covered by current vendor catalogs are only a fraction of the available chemical space, standard virtual screening approaches may not always identify molecules optimal for the specific protein binding site being targeted.
Alternative methods, such as de novo design (hereafter abbreviated as DN), provide complementary ways to navigate and search chemical space using algorithms that enable molecules to be constructed from-scratch, directly in the context of the binding site,5–11 thereby eliminating the need to start from libraries of pre-existing compounds. In theory, DN approaches should allow users to identify target-compatible compounds more quickly, and with less computational effort, than with virtual screens. In practice, molecules constructed using DN may have shortcomings that need to be addressed. For example, although DN molecules may have been constructed to interact favorably with the drug target, they may also be difficult to synthesize chemically,12 or have unfavorable cheminformatic properties typically required of drug-like candidates. Recently, several studies have employed artificial intelligence and/or machine learning in an attempt address some of these known shortcomings.13–18 In our own work, we employed data mining to develop an “allowable torsion type” table to enforce chemically reasonable growth during DN, based on deconstruction of 13M drug-like molecules, so that only previously observed torsion types would be allowed. A conceptually similar strategy, presented in the paper outlining the program OpenGrowth,19 employed functional group pairwise probabilities (termed FOG probabilities) to help promote physically reasonable molecules.
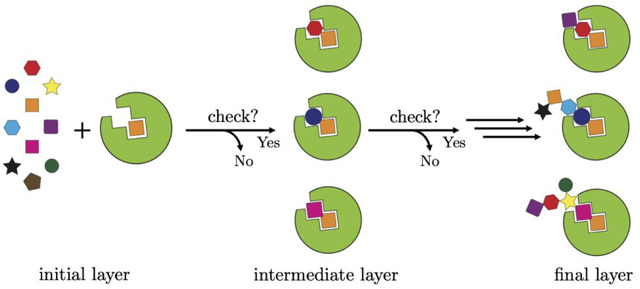
In the present work, we describe a significant new addition to the DOCK6 de novo design engine (DOCK_DN) that builds upon prior work described in Allen et al.20 As shown in Figure 1a, ligand assembly in DOCK_DN begins with the orientation of a molecular fragment (termed anchor) selected from a user definable fragment library (see Methods). Candidate fragments are then attached to each anchor, in a layer-by-layer fashion, similar to the standard DOCK6 anchor-and-grow algorithm,21 until a complete molecule is obtained. We hypothesized we could improve upon the current DOCK algorithms by checking whether partially grown molecules have reasonable drug-like properties at each layer of DN growth (Figure 1, horizontal arrows) using cheminformatics descriptors such as lipophilicity, solubility, topological polar surface area, or ease of synthesis, among others. The objective is to construct more pharmacologically favored ensembles, directly in the protein binding site, without the need for post-processing or filtering. We call our methodology “Descriptor Driven De Novo” (abbreviated D3N) because the decision to promote partially grown molecules to the next layer of growth is contingent on user-defined ranges for the descriptor being employed. The primary objective of the present study is three-fold: (1) implement and validate a robust interface enabling DOCK6 to communicate with the open-source cheminformatics package RDKit,22 (2) confirm that the use of DOCK_D3N protocols leads to ligand ensembles that conform to the desired target values for the descriptors under different conditions and environments, and (3) examine ligand growth behavior using very narrow ranges for descriptors derived from clinically-relevant compounds.
Figure 1.

Schematic outlining showing the D3N algorithm. (1a) An anchor (initial layer, orange square) is selected from the fragment library (colored fragments) and oriented/scored in the protein binding site (green). (1b) As candidate fragments are added to the anchor, the partially grown molecules must conform to the user-defined descriptor ranges (intermediate layer, orange + colored fragment) or the fragment is rejected. The process continues until the desired number of layers is reached (the present work employed up to 9 layers of growth). (1c) The final ensemble will be enriched with fully grown molecules (multi-colored and connected fragments) that conform to the user-defined descriptor ranges.
Computational Methods and Details
Software interface and infrastructure.
The DOCK623–25 program is written primarily in C++ and the most recent version (DOCK6.10) has three main engines for ligand sampling and chemical searching: (1) virtual screening using an anchor-and-grow algorithm,21 (2) from-scratch construction using a DN design algorithm (DOCK_DN),20 and (3) molecular evolution using a genetic algorithm (DOCK_GA).26 The primary objective of the present work was development and testing of a DOCK6/RDKit interface to allow cheminformatics descriptors to be used in conjunction with DOCK_DN.20 This required adapting the DOCK6 object DOCKMol to communicate with RDKit objects ROMol and RWMol. The names of relevant functions and input parameters in this manuscript are highlighted in italics. Briefly, the new DOCK6/RDKit interface, named DOCKMol_to_ROMol, assigns DOCKMol object atom types, bond orders, formal charges, and other molecular properties to ROMol that is ultimately used to calculate the RDKit descriptors. DOCKMol_to_ROMol is only used in circumstances when descriptors are required otherwise the standard DOCKMol object is employed. The DOCKMol_to_ROMol interface was largely inspired by the Mol2FileParser routine provided with the standard RDKit distribution.
The RDKit interface for DOCK in this work was developed on top of DOCK6.9 (dock.compbio.ucsf.edu) using the 2019.09.01 release of RDKit (rdkit.org) and Boost 1.71.0 (boost.org). The RDKit compilation process also requires Boost 1.71.0 (boost.org) in addition to Eigen 3.3.9 (eigen.tuxfamily.org) and Anaconda3 (anaconda.com). All code was compiled with GNU compilers (gcc/g++ 7). DOCK6 users have a choice if they want to compile DOCK6 with RDKit. If so, they will need to add the path for RDKit and Boost to their bashrc (or equivalent) file. In principle, the interface will allow any of the > 50 RDKit descriptors (2D, 3D, fingerprints, and combinations thereof) to be used by DOCK6. This current work has focused on interfacing a relatively small subset of key descriptors including QED (quantitative estimate of druglikeness),27 SynthA (synthetic accessibility),28 TPSA (topological polar surface area),29 LogP (octanol/water partition coefficient),30 LogS (aqueous solubility),31 #Aromatic (number of aromatic rings), #Stereo (number of stereocenters), #Spiro (number of spirocenters), #PAINS (number and identity of pan-assay interference compounds),32–34 #Aliphatic (number of aliphatic rings), #Saturated (number of saturated rings), MACCS fingerprint keys,35 and SMILES strings. While most of these 13 descriptors could be interfaced immediately, QED, SynthA, and LogS required conversion of Python scripts in the RDKit Github repository to C++. A new DOCK6 class, termed RDTyper, was written to retrieve RDKit descriptors, and perform calculations using “combinations of descriptors” as needed. RDtyper can be called from the DOCK6 database filter/utilities class23 or the DOCK_DN engine.20 Readers should note that current D3N protocols allow 7 of the aforementioned 13 descriptors to be used during on-the-fly growth. Future work will evaluate use of other descriptors for D3N, as well as develop an RDKit interface for the DOCK6 genetic algorithm (DOCK_GA) recently reported by Prentis et al.26
D3N fragment library, algorithm, and implementation.
As illustrated in Figure 1, docked (oriented) ligand fragments (termed anchors) are used to seed DN growth followed by the attachment of compatible fragments (i.e. allowable newly-formed torsion types), one-by-one, over the course of a user-defined number of steps (typically 8–9). Figure 2a visually illustrates how fragment libraries are constructed using DOCK6 starting from a collection of input molecules, in this example epinephrine and DANA. Figure 2b shows 3D representations for 19 fragments, ordered by frequency of occurrence, derived from deconstructing 13,195,579 drug-like molecules downloaded from ZINC.2–4 As in prior work,20,26 for tractability, we choose to retain molecular fragments only if they appeared 13,000 times or greater (roughly ~0.1% of the total) resulting in a final curated set of 382 fragments and 10,844 allowable torsions (bond types). For consistency with prior work all allowable torsion types were retained. The library is arranged into sidechains (1 attachment point, N=217), linkers (2 attachment points, N=146), and scaffolds (3+ attachment points, N=19). It is important to note that the DOCK6 infrastructure allows users to easily customize their own fragment libraries and retain as many entries (fragments or torsion types) as desired.
Figure 2.

Schematic illustrating (a) how DOCK6 fragment libraries are derived by deconstructing molecules along rotatable bonds and (b) the top 19 fragments ordered by frequency (out of 382 retained) for a library derived from 13M drug-like molecules downloaded from ZINC.2–4 Based on the number of dummy atoms (magenta attachment points) the fragments are classified into sidechains (1 attachment point, N=217), linkers (2 attachment points, N=146), or scaffolds (3+ attachment points, N=19).
At each stage of DN growth, multiple 3D geometries are generated (sampling), the fitness of the partially grown molecule is evaluated (scoring), and a small number of molecular properties are computed including molecular weight, number of rotatable bonds, formal charge, number of potential H-bond acceptors, and number of potential H-bond donors. The new D3N algorithm adds to these features by allowing up to 7 additional descriptors to influence ligand growth including QED (dn_drive_qed), SynthA (dn_drive_sa), TPSA (dn_drive_tpsa), LogP (dn_drive_clogp), LogS (dn_drive_esol), #Stereo (dn_drive_stereocenters), and #PAINS (dn_drive_pains). Each descriptor can be turned on or off with the user having full control over the target ranges (discussed below). As each partially grown molecule is passed to the next layer of growth (Figure 1), if the values for computed descriptors fall within the target ranges defined in the input file, the molecule is automatically stored in a separate vector to be sent to the next layer. If any descriptor falls outside of the target range, the acceptance of the molecule is determined by a Metropolis-like procedure (termed soft-cutoff scheme) as shown in Figure 3. Here, probability of acceptance is calculated assuming a normal distribution of descriptor values where is the descriptor value, is the nearest interval limit, is the descriptor standard deviation (see next section), and is a random number between 0 and 1. If, the molecule is accepted and sent to the next layer. If the molecule is rejected. Readers should note that #Stereo and #PAINS have a slightly different criterion that takes into consideration their discrete (integer) values according to Eq. 1 where N is an integer that should always be greater than 1 during application of the soft-cutoff scheme.
Figure 3.

Schematic showing D3N procedure for accepting new fragments. For every molecule at each layer of growth, multiple descriptors are calculated. If all descriptors fall within the user-defined ranges, the partially-grown molecule is accepted and sent to the next layer of growth. If one or more descriptors are outside the target range, a soft-cutoff (Metropolis-like) scheme is applied in which there is a finite probability that the molecule could be sent to the next layer.
| (EQ. 1) |
Simulation types (simple-build, protein-standard, protein-pinpoint).
The primary simulation types employed in this work, shown in Table 1, can be organized into three broad categories: (1) growth in the absence of a protein site termed “simple-build”, (2) growth in protein sites using standard parameter ranges termed “protein-standard”, and (3) growth in protein sites with very specific parameters termed “protein-pinpoint”. While the different simulation types employed different numbers of anchors as seeds for growth, in subsequent layers, all 382 fragments were employed. Readers should note that as used here, anchors are subset of the total number of fragments. Table 1 also lists the pdb codes for each protein or protein family and the main scoring function(s) employed in each case.
Table 1.
Primary simulation types employed for D3N growth.
| Simulation Type | N Anchors, N Fragments | Main Scoring Function Employed |
|---|---|---|
| (1) Simple-build (absence of protein) | 380, 382 | Ligand only VDW repulsive term |
| (2) Protein-standard (57 systems) a | 15, 382 | Single Grid Energy (SgE) |
| (3) Protein-pinpoint (6 systems) b | 380, 382 | Multi Grid Energy (MGE) + Footprint Similarity (FPS) |
acetylcholinesterase (1EVE, 1H22, 1J07, 1Q84, 1ZGC), cyclooxygenase (1EQG, 1EQH, 1HT5, 1HT8, 1Q4G, 4COX), EGFR (2ITP, 2ITT, 2ITY, 2RGP, 3BEL), HIV protease (1AJV, 1DMP, 1HVR, 1MER, 1MES, 1MET, 1QBS, 2F80, 2F81, 2IDW, 2IEN, 2IEO), HIV reverse transcriptase (1C1B, 1C1C, 1VRU, 2BE2, 2RKI, 2ZD1, 3BGR, 3DLE, 3DLG, 3DOL), IGF1R (2ZM3, 3NW5, 3NW6, 3NW7), neuraminidase (1BJI, 1F8B, 1F8C, 1F8D, 1F8E, 1MWE, 1NNB, 1NNC, 1XOE, 1XOG), streptavidin (1DF8, 1SRG, 1SRI, 1SRJ, 2IZL).
MAP kinase (1A9U), FGR1 kinase domain (1AGW), COX-1 (1EQH), HMG-CoA reductase (1HW9), HIV reverse transcriptase (1IKW), neuraminidase (3CL0).
Growth under the simple-build infrastructure is guided by a ligand-only energy function comprised solely of an intramolecular van der Waals (VDW) repulsive term (Lennard Jones coefficient of 12). Given that simple-build results are not influenced by binding site characteristics, this helps to isolate behavior of the algorithm. Simple-build is also significantly faster than protein-based simulations thus we employed nearly all of the fragments in the library as anchors to seed growth (N=380) which increases chemical searching.
Growth under the protein-standard infrastructure (N=57, eight protein families) is guided by the standard dock single grid energy (SGE) function comprise of non-bonded VDW plus electrostatic (ES) interactions. However, given the increased computational expense and the large number of protein systems involved, only the first 10 most commonly occurring fragments in Figure 2 were used as anchors (#1–10) augmented by 5 less frequently occurring fragments chosen at random to explore different chemistries: triazole (#100), adamantyl (#250), thiazole (#300), sub-chlorobenzene (#350), and sub-oxazole (#380).
Growth under the protein-pinpoint infrastructure (N=6 systems) used an enhanced scoring function comprised of multi-grid (MGE) plus footprint similarity (FPS) terms (FPS weights = 1). The addition of FPS36,37 scoring helps bias growth towards the interaction signatures made by a reference. In this work, the ligand bound to each of the 6 crystal structure targets served as the reference and are referred to in the remainder of the manuscript as the “reference ligand”. To facilitate energetic comparisons between the reference ligands and outcomes from de novo design, the references were assigned hydrogen atoms, Gasteiger-Marsili charges,38 and minimized in the same multi-grids employed during D3N growth. Here, given the smaller number of pinpoint systems examined, the larger group of anchors (N=380) was used.
System setup details for protein-based simulations.
Protein simulations in this work employed setups taken from our SB2012 dataset, the construction of which has been previously described.23,39 20 Briefly, for each system, coordinates files were downloaded from the PDB40 and saved as separate protein and ligand entries. Ligands were protonated, visually examined for correctness, and assigned partial atomic charges (AM1-BCC method)41,42 and force-field parameters (GAFF)43 using the program antechamber44 distributed with the Amber45 suite of programs (AmberTools). Amber-ready protein-ligand complexes were then assembled with the program tLEaP (AmberTools) which protonates the protein and assigns ff99SB46 parameters. After preparation, each complex was energy minimized using Amber1647 (using heavy atom restraints) to relax the system with the force field and alleviate any potential clashes that might have arisen from the addition of hydrogen atoms. The minimized protein was extracted, saved in MOL2 format, and used as input for the program DMS48 (1.4 Å radius probe) to generate a molecular surface. In turn, the DMS surface is used as input to the program sphgen49 to generate a set of docking spheres used to orient anchors (or ligands) in the binding site. Finally, the DOCK accessory program GRID50 was used to create a set of docking grids, which speed up the calculations, by pre-computing VDW (6–9 attractive-repulsive Lennard-Jones exponents) and ES (Coulombic interactions scaled by a distance-dependent dielectric = 4𝑟) contributions from the protein.
DOCK_DN simulation parameters.
The key DOCK_DN parameters employed in this work are listed in Table 2. For additional information, readers should consult Allen et al20 and the DOCK6 manual (dock.compbio.ucsf.edu). Briefly, all simulations employed the random sampling method (dn_sampling_method) for which up to 20 or 50 (protein-pinpoint simulations only, see discussion above) fragment selections (picks) were allowed (dn_num_random_picks). In addition to the D3N-specific parameters discussed above, additional constraints (see Table 2 for values) evaluated during growth included filters for molecular weight (dn_mol_wt_cutoff_type, dn_upper_constraint_mol_wt, dn_lower_constraint_mol_wt, dn_mol_wt_std_dev), number of rotatable bonds (dn_constraint_rot_bon), and formal charge (dn_constraint_formal_charge). The maximum number of growth layers per molecule (dn_max_grow_layers) was set to 9, the maximum number of molecules that can be derived from any single partially grown molecule or “root” (dn_max_root_size) was set to 25 or 50 (protein-pinpoint only), and the maximum ensemble size of partially grown molecules that can be passed to the next layer (dn_max_layer_size) was set to 25 or 50 (protein-pinpoint only). The maximum number of scaffold fragments that could be added to any molecule at each layer of growth (dn_max_scaffolds_per_layer) was capped at 1 and the maximum number unsatisfied attachment points per molecule (dn_max_current_aps) at any point was set to 5 which helps control branching.20
Table 2.
Key DOCK_DN parameter values used in this work.
| Parameter | Description | Value |
|---|---|---|
| dn_sampling method | Method employed for picking fragments (exhaustive, random, graph) | random |
| dn_num_random_picks | N fragments randomly selected | 20, 50 |
| dn_mol_wt_cutoff_type | Molecular weight filtering method (hard, soft) | soft |
| dn_upper_constraint_mol_wt | The upper limit for MW filter | 550 |
| dn_lower_constraint_mol_wt | The lower limit for MW filter | 0 |
| dn_mol_wt_std_dev | The standard deviation for MW filter | 35 |
| dn_constraint_rot_bon | The max rotatable bonds allowed | 15 |
| dn_constraint_formal_charge | Largest absolute charge of molecule | 2 |
| dn_max_grow_layers | Max number of layers for growth starting from an anchor | 9 |
| dn_max_root_size | Max number of new molecules allowed from any given growing molecule | 25, 50 |
| dn_max_layer_size | Max number of partially grown molecules that advanced to the next layer | 25, 50 |
| dn_max_current_aps | Max number of unsatisfied attachment points at any given time | 5 |
| dn_max_scaffolds_per_layer | Max number of scaffolds added per layer per molecule | 1 |
As noted previously,20,26 chemical searching in DOCK_DN can lead to molecules with identical topology but different conformations and/or binding poses. In the present work, to simplify interpretation of D3N outcomes, “duplicate” molecules were removed by (1) grouping all molecules for a given experiment into single MOL2 file, (2) clustering the molecules based on topological identity using SMILES strings, and (3) retaining only those molecules with the best score depending on the experiment (internal energy, single grid energy, or multi-grid energy).
Default descriptor ranges derived from approved small molecule drugs and active pharmaceutical agents.
For testing D3N, we wanted a reasonable set of input parameter ranges (DOCK6 defaults) for the 7 different descriptors. Table 3 shows values derived from a curated set of approved small molecule drugs and active pharmaceutical agents contained in the DrugCentral database,51,52 termed here the “D3N-drugc” parameter set (see Supporting Information for curation details), based on values computed using DOCK6/RDKit (Table S1). The parameter ranges for TPSA, LogP, and LogS reflect the mean ± the standard deviation from corresponding entries in Table S1 (Supporting Information). For QED, SynthA, #Stereo, and #PAINS, one-sided boundary ranges were employed given that the “best” scores have direction. For example, QED scores range from 0 to 1 with 1 being best.27 Conversely, SynthA scores range from 1 to 10 with 1 being best. For these two descriptors, D3N-drugc default values were set to their respective DrugC means of 0.61 (QED, lower bound only) and 3.34 (SynthA, upper bound only).28 For #Stereo and #PAINS, given that scores near 1 or 0 would likely be desirable, upper bound values for pruning were set to 2 and 1, respectively. Table 3 also contains values for a protocol termed “D3N-loose”, meant to mimic standard DN behavior (little to no RDKit-based pruning), which provides a control.
Table 3.
Input parameters ranges for D3N-drugc and D3N-loose protocols.
| Descriptor name | D3N-drugc a range | D3N-drugc std dev | D3N-loose b range |
|---|---|---|---|
| QED | 0.61 lower bound | 0.19 | 0.0 lower bound |
| SynthA | 3.34 upper bound | 0.90 | 10 upper bound |
| TPSAc | 28.53 to 113.20 | 42.33 | 0 to 9999 |
| LogP | −0.30 to 3.75 | 2.02 | −20 to 20 |
| LogS | −5.23 to −1.35 | 1.94 | N/A |
| #Stereo | 2 upper bound | N/A | 100 upper bound |
| #PAINS | 1 upper bound | N/A | N/A |
D3N-drugc parameter ranges mimic DrugC dataset distributions (± one std dev from mean).
D3N-loose parameter ranges mimic standard DOCK_DN behavior (little to no pruning).
TPSA values in angstroms squared.
Descriptor correlations.
Although the DOCK6/RDKit interface allows multiple descriptors to be employed simultaneously during ligand growth, it is important to assess the extent with which different descriptors may be correlated. D3N simulations employing non-orthogonal descriptor combinations may suffer from sampling issues and inefficient navigation of chemical space. Figure 4 shows a Pearson matrix correlation heatmap from pairwise combinations of the five “non-integer” D3N descriptors using molecules from ZINC13M. As shown by the heatmap, the descriptors show relatively weak correlation with the exception of LogP and LogS which yielded a R value of −0.96 (strong inverse correlation). To avoid multicollinearity, we opted not to drive LogP and LogS simultaneously.
Figure 4.

Pearsons correlation matrix between descriptors computed for molecules in the ZINC13M dataset color-coded as a heatmap.
Results & Discussion
Code and infrastructure validation.
To establish that the DOCK6/RDKit integration was implemented correctly, we computed 9 descriptors for the 13M molecules in the ZINC13M dataset and compared the numerical results with those obtained using the standard Python3 RDKit distribution (Python/RDKit). To establish a computationally consistent environment, the SMILES strings generated internally using DOCK6/RDKit were also used as the input for Python/RDKit. As shown in Figure 5, the numerical results from both platforms are identical, for all practical purposes, which confirms the integrity of the implementation. Some minor exceptions are observed in plots of QED, SynthA, and LogS which are numerically insignificant. Readers should note that the heatmap colors in Figure 5 are a complementary way to visualize the descriptor populations shown in Figure S1 which compare the underlying descriptor distributions for the DrugC and ZINC13M datasets.
Figure 5.

Scatter plots for descriptors calculated using DOCK6/RDKit (DOCK6_RDK) vs Python/RDKit (Python_RDK) using 13M molecules downloaded from ZINC (ZINC13M dataset). Both sets of calculations employed the identical SMILES strings generated using DOCK6/RDKit from MOL2 files. Heatmap colors correspond to the number of molecules (population) across each descriptor range. TPSA values in angstroms squared.
It should be emphasized that the numerically equivalent results in Figure 5 comparing DOCK6/RDKit to Python/RDKit are a direct result of using the identical SMILES strings as inputs for both sets of calculations. Importantly, they establish that the underlying calculations methods are the same. However, DOCK6/RDKit requires MOL2 files as input, for which SMILES strings are generated internally prior to the calculations, and Python/RDKit users typically employ SMILES as input. To help gauge the accuracy of the MOL2 to SMILES conversion routine we performed additional testing using a large curated set of compounds derived from ZINC15 (N=11,292,054) for which both a MOL2 file and a SMILES string were available for each ZINCID. Compounds were downloaded via the ZINC15 tranche browser subject to the following criteria: MW = 300 to 500, LogP = 0 to 5, formal charge = 0, and pH = Ref. In an attempt to mitigate any obvious changes that could lead to numerical differences arising from different protomers or tautomers we eliminated entries from the downloaded tranches if: (1) a given ZINC IDs had multiple SMILES strings, (2) a SMILES string and its associated MOL2 file contained a different number of hydrogen atoms, or (3) uncommon elements were present (e.g. silicon and metals).
Reassuringly, as shown in Figure 6, for nearly 100% of the 11M molecules evaluated, the identical numerical results were obtained across all 9 descriptors independent of whether a MOL2 file (DOCK6_RDK_mol2) or SMILES file (Python_RDK_smi) was used as the input. This confirms the reliability of the DOCK6/RDKit MOL2 to SMILES conversions. Interestingly, despite our best efforts at pre- and post-filtering ZINC15, a cursory examination using the ZINC15 web browser showed that for several outliers the MOL2 and SMILES forms labeled as pH = Ref and charge = 0 had different tautomeric states. In some cases, there also appeared to be differences in resonance state or number of implicit bonds. In a practical sense, this suggests that users should be careful to ensure that the correct tautomeric states are represented as desired when computing descriptors using either input format (MOL2 or SMILES).
Figure 6.

Scatter plots for descriptors calculated using DOCK6/RDKit with SMILES generated from MOL2 files (DOCK6_RDK_mol2) vs Python/RDKit with SMILES directly from ZINC (Python_RDK_smi). Heatmap colors correspond to the number of molecules (population) across each descriptor range. TPSA values in angstroms squared.
Descriptor driven de novo design (D3N) of ligands in the absence of a protein.
Single descriptor design (D3N-lateral protocol) dramatically shifts distributions.
Having validated that the DOCK6/RDKit implementation is robust, we next assessed the ability of our descriptor driven de novo design (D3N) algorithm to generate new ligands using the “simple-build” infrastructure (absence of protein) which speeds up calculations. Figure 7 shows the distributions for three descriptors (SynthA, TPSA, LogP) derived from molecules grown using a protocol in which only a single descriptor at a time was employed for pruning at every layer of growth. Growth here was based on setting the target D3N ranges for each descriptor aggressively (Figure 7, target ranges in red font) in that they were laterally shifted left or right (termed D3N-lateral, red) relative to those obtained with the D3N-loose (gray) protocol (little to no pruning control). The accompanying values in parenthesis in Figure 7 specify how many molecules were generated in each case. Results are separated into those (a) employing the “standard” fragment library supplied with DOCK6 (382 fragments, 10,844 torsions) or (b) an alternative “focused” fragment library (389 fragments, 603 torsions) derived from molecules with negative LogP values.
Figure 7.

Normalized descriptor populations using (a) the standard DOCK6 fragment library or (b) a focused fragment library derived from molecules with negative LogP values. In each case, a single descriptor with laterally shifted target ranges (D3N-lateral, red) were used to drive de novo growth in the absence of protein. Results obtained using the D3N-loose protocols (gray) are shown as a control. Legends indicate the specific target ranges (labeled in red font) and the number of molecules obtained (in parenthesis). Readers should note this data was derived from four independent experiments. The standard deviations employed for D3N-lateral protocols are the same as listed in Table 3 for D3N-drugc. TPSA results in angstroms squared.
Importantly, in each case, use of the D3N-lateral protocol (red) yield distributions that are shifted towards their intended target range (Figure 7 red labels) and they are more tightly focused which provides evidence that the D3N infrastructure is working as intended. As expected, in all cases, on-the-fly D3N pruning leads to fewer molecules being produced. For SynthA (Figure 7a left), which employs a one-sided boundary, the initially broad D3N-loose distribution (gray, 2 to 6) becomes tightly focused with the D3N-lateral protocol (red, 1 to 4) and the resulting left-shifted peak (~3) is close to the intended target (upper range 2). For TPSA (Figure 7a middle), which employs a two-sided boundary, good agreement is also obtained. Here, D3N-lateral (red) results shift right, which nicely spans the intended TPSA target range (150–250), and the distribution peak (~170) is near the center (200). For LogP however (Figure 7a right), although the D3N-lateral (red) results are significantly more focused, and correctly shift left towards the target range (−10 to 0), the ensemble contained very few molecules that extended below −2.5 and the peak was not near the range center (−5).
The absence of designed molecules with negative LogP was somewhat surprising, which prompted us to more closely examine molecules in the DrugC dataset. De novo design outcomes depend on many factors, including fragment libraries, and many molecules in the DrugC dataset with negative LogP contained functionality not present in our standard DOCK6 fragment library, for example, phosphates, tetracycline rings, or beta-lactam fused rings. We hypothesized that an alternative library, containing such fragments, would lead to assemble of compounds enriched for negative LogP. Figure 7b plots D3N-lateral results using an alternative “focused” library comprised of 398 fragments and 603 allowable torsions which was derived from the disassembly of 494 molecules in the DrugC dataset with negative LogP values. Reassuringly, the experiment showed a large increase in the number of molecules with negative LogP (Figure 7b vs 7a) for both the D3N-lateral protocol (57.05 vs 20.21%, red) and the D3N-loose control protocol (33.46 vs 5.30%, gray). The test also provides context for any given computational protocol being able to achieve a desired descriptor “range” given the sensitivity of de novo design to the composition of the fragment libraries. They also establish that D3N protocols can be used to shift descriptor distributions regardless of the libraries employed. Notwithstanding the importance of including specific fragment types that may lead to more negative LogP values (or other ranges for other properties), for the remainder of the tests in this manuscript, the standard DOCK6 fragment library was employed.
Multi-descriptor design (D3N-drugc protocol) focuses multiple descriptors simultaneously.
In a second group of “simple-build” experiments (absence of protein), we evaluated the ability to drive multiple descriptors simultaneously employing ranges derived from molecules in DrugCentral (D3N-drugc protocol, Table 3). Here, although DOCK6 can currently compute 13 RDKit descriptors, of which 7 are available for on-the-fly de novo design, we opted not to drive LogS (to avoid multicollinearity, see Figure 4) or #PAINS (initial tests showed we rarely generate PAINS molecules using the standard fragment library). Thus, the final group of 5 descriptors employed included QED, SynthA, TPSA, LogP, and #Stereo. Figure 8 compares distributions obtained using the multi-descriptor D3N-drugc protocol (solid red, pruning at every layer) with D3N-loose (gray, little to no pruning) and the DrugC dataset (dashed red). As before, accompanying values in parenthesis specify how many molecules were generated using each protocol.
Figure 8.

Outcomes from multi-descriptor de novo design using D3N-drugc (solid red) protocols driving QED, SynthA, TPSA, LogP, and #Stereo simultaneously compared to D3N-loose (gray) as a control. The distributions for molecules in the DrugCentral data set (dashed red) are shown for comparison. Multi-descriptor target ranges listed in Table 3. Values in parenthesis specify how many molecules were generated with each protocol. TPSA values in angstroms squared.
As shown in Figure 8, compared to using the D3N-loose (gray) protocol, the multi-descriptor D3N-drugc protocol (solid red) yields molecules with distributions that are in general more focused (tighter) and shift left or right in the direction of their intended target ranges (DrugCentral distributions, dashed red). This demonstrates that descriptor-based pruning can be used to drive multiple descriptors simultaneously. For QED, TPSA, and to a lesser extent LogP, the peaks for D3N-drugc (solid red) land in-between those of D3N-loose (gray) and the DrugCentral dataset (dashed red). QED shows a large shift from a single peak at around 0.25 (D3N-loose, gray, poor druglikeness) to a bimodal shape with two peaks between 0.5 and 0.7 (D3N-drugc, solid red, higher druglikeness). Although not explicitly driven, the D3N-drugc distributions (solid red) for LogS and #Aromatic rings also show significant shifts towards DrugCentral (dashed red) likely as a result of descriptors coupling arising from the strong anti-correlation between LogS and LogP (−0.96, Figure 4) and #Aromatic being a key component of the QED scoring function (additional discussion below).
Single descriptor design can influence multiple descriptors.
Of the nine descriptors in Figure 8, the distributions for QED standout as having the overall poorest agreement between D3N-loose results and the DrugCentral dataset (Figure 8 gray vs dashed red). QED is a combination of multiple descriptors including TPSA, LogP, and #Aromatic in conjunction with MW, #H-bond donors, #H-bond acceptors, number of rotatable bonds, and number of structural alerts.27 In a third set of “simple-build” experiments (Figure 9), we wanted the extent with which driving QED alone would show a concomitant change in some of the underlying descriptors that make up the total score. We also wanted to examine the behavior of applying the D3N algorithm at different layers of growth. We hypothesized that initiating D3N in early layers would lead to a cumulative effect in term of more closely matching the intended target range. Figure 9 plots results from driving QED alone (starting at layer 1, 5, or 9) versus driving QED, SynthA, TPSA, LogP, and #Stereo simultaneously (starting at layer 1).
Figure 9.

De novo design outcomes in the absence of protein (simple-build protocol) using single descriptor D3N-drugc protocols to only drive QED as a function of which growth layer the pruning algorithm takes effect (layer 1 red, layer 5 purple, layer 9 blue). Results from the multi-descriptor D3N-drugc protocol are plotted for comparison (pink shaded areas). TPSA values in angstroms squared.
As shown in Figure 9a, driving QED alone starting at layer 1 (D3N-drugc 1 single, red) yields a QED distribution which largely mimics the shape from the multi-descriptor distribution (red vs pink shade). And, the distributions for some of the individual terms that make up QED (there are eight terms total), including TPSA (Figure 9b), LogP (Figure 9c) and #Aromatic (Figure 9d) show a concomitant change, even though they were not specifically pruned to do so, that also approaches the multi-descriptor distributions (Figure 9b-d, red vs pink). Importantly, as the D3N algorithm becomes initiated earlier in the process the distributions become progressively focused in a relatively smooth manner (layer 9 blue to layer 5 purple to layer 1 red). Overall, the data in Figure 9 provide additional evidence that the D3N algorithm is well-behaved, demonstrate that changes in a given descriptor distribution can be coupled to driving other descriptors, and show that driving QED alone with a single-descriptor protocol leads to outcomes approaching that of a multi-descriptor protocol.
Descriptor driven de novo design (D3N) of ligands in protein binding sites.
Multi-descriptor design in proteins yields focused distributions.
The three different D3N simple-build experiments, described above, demonstrate that it is possible to bias chemical searching to regions defined by the user in the absence of a protein. This is to be expected, given that the pruning is primarily driven by the descriptors themselves, although other properties are also at play (i.e. newly formed bonds must be allowable and other properties including MW, number of rotatable bonds, and formal charge, among others in Table 2, must be met). In this section, we evaluate the ability to generate drug-like candidates in clinically-relevant protein targets taken from our SB2012 docking database.23,39 In contrast to simple build, D3N growth in a protein will be heavily influenced by the protein-ligand interaction energy. Growth here was seeded in 57 individual binding sites structures (see Table 1 for individual pdb codes) comprised of 8 protein families: acetylcholinesterase (5 systems), cyclooxygenase (6 systems), EGFR (5 systems), HIV protease (12 systems), HIV reverse transcriptase (10 systems), IGF1R (4 systems), neuraminidase (10 systems), and streptavidin (5 systems). The protein environment simulations take significantly longer than the previous simple-build experiments thus only 15 fragments (see Methods) were used to seed ligand growth in each of the 57 systems.
Figures 10 and 11 show descriptor distributions for QED, SynthA, TPSA, LogP (Figure 10 top) and #Stereo (Figure 11) driving all five descriptors simultaneously using the same D3N-drugc (red) or D3N-loose (gray) ranges employed in the earlier simple-build experiments (Table 3). Results for a third protocol, termed D3N-narrow (purple), are shown in Figure 10 to highlight the ability of the algorithm to generate narrow distributions (target ranges shown above each plot). As expected, in every case, the distributions from D3N-drugc (red) and D3N-narrow (purple) are more focused relative to D3N-loose (gray). The D3N-narrow results (purple) in particular show extreme focusing (Figure 10 top, purple) and yield peak locations in close agreement with the intended target ranges listed above each plot. As shown in Figure 11, use of a #Stereo upper target range of 2 with the D3N-drugc protocol (0.93%, red) leads to very few molecules with more than two stereocenters compared to the D3N-loose protocol (14.1%, gray).
Figure 10.

QED, SynthA, TPSA and LogP distributions obtained using D3N-drugc (red), D3N-loose (gray shade) and D3N-narrrow (purple) in 57 protein binding sites starting from 15 fragments as anchors. D3N-drugc and D3N-loose target ranges listed in Table 3. D3N-narrow target ranges shown in purple above each plot. Bottom panels compare distributions for the D3N-drugc rejected (dashed red) and accepted (solid red) molecules. Distributions obtained by kernel density estimation. TPSA values in angstroms squared.
Figure 11.

Histogram populations for #Stereo (number of ligand stereocenters) using D3N-drugc (red) or D3N-loose (gray) simulation protocols in 57 protein binding sites.
To further probe the algorithm, Figure 10 (bottom) plots descriptor distributions using the D3N-drugc protocol based on the rejected (dashed red) or accepted (solid red) molecules which confirms the Metropolis-like criteria is being obeyed with regards to enforcing soft-cutoff boundaries (see Figure 3). For TPSA and LogP, which have two-sided boundaries, the rejected molecule profiles show clear bimodal distributions (dashed red) spanning opposite sides of the accepted molecule profiles (solid red). Table 4 shows the number of unique molecules generated via the three different protocols. As expected, the construction trend in terms of number of molecules follows D3N-loose (282,989) > D3N-drugc (184,118) > D3N-narrow (11,903) and the trend for rejection is in the opposite order with D3N-loose (724) < D3N-drugc (565,823) < D3N-narrow (651,423). Taken together, these results confirm the ability of the algorithm to successfully create and prune molecules, based on user-defined ranges for descriptors computed using the DOCK6/RDKit interface, for de novo design performed in the context of a protein binding site.
Table 4.
Number of unique molecules constructed using different D3N protocols in 57 proteins starting from 15 different fragments each as anchors for growth over 9 layers.
| D3N-loose | D3N-drugc | D3N-narrow | |
|---|---|---|---|
| Constructed a | 282,989 | 184,118 | 11,903 |
| D3N-rejected | 724 | 565,823 | 651,423 |
Values reflect the number of unique molecules created for each anchor simulation with duplicates entries removed (see Methods).
On-the-fly pruning leads to molecules enriched for favorable properties.
As shown thus far, the DOCK6/RDKit implementation allows users to prune unwanted molecules during the de novo design process but it can also be used as a post-processing filtering tool. To help gauge the added value for using on-the-fly pruning, versus post-simulation filtering, the ensembles created using D3N-loose and D3N-drugc protocols were both processed by applying hard-cut filtering to remove molecules at the boundaries arising from use of the soft-cutoff algorithm. The two datasets were filtered using the same five property cutoffs (D3N-drugc ranges in Table 3) resulting in 39,159 molecules for D3N-loose (from 282,989 raw) and 44,419 molecules for D3N-drugc (from 184,118 raw) as shown in Table 5. From an enrichment standpoint, the filtered D3N-drugc protocol yielded an additional 5,260 molecules which is a 13.43% increase across all 57 systems. Encouragingly, enrichments for all the protein families with the D3N-drugc protocol generated more molecules ranging from 4.87% to 25.56% (Table 5, far right column). Figure 12 also highlights that the filtered ensembles from D3N-drugc (red) contain more molecules than the filtered ensembles from D3N-loose (gray) in terms of favorable QED and SynthA scores, and within the target ranges specified for TPSA and LogP.
Table 5.
Number of molecules from simulations using D3N-loose and D3N-drugc protocols.
| protein family (No.) | D3N-loose rawa | D3N-drugc rawa | D3N-loose filteredb | D3N-drugc filteredb | Relative increase for filtered (drugc vs loose)c | |
|---|---|---|---|---|---|---|
| acetylcholinesterase (5) | 26,079 | 16,310 | 2,954 | 3,699 | 745 | 25.22% |
| cyclooxygenase (6) | 19,237 | 13,823 | 4,004 | 4,425 | 421 | 10.51% |
| EGFR (5) | 25,910 | 15,956 | 3,571 | 3,745 | 174 | 4.87% |
| HIV protease (12) | 71,480 | 45,069 | 7,484 | 8,637 | 1,153 | 15.41% |
| HIV reverse transcriptase (10) | 44,793 | 30,161 | 8,440 | 8,985 | 545 | 6.46% |
| IGF1R (4) | 22,325 | 14,227 | 2,903 | 3,165 | 262 | 9.02% |
| neuraminidase (10) | 49,987 | 33,671 | 6,111 | 7,673 | 1,562 | 25.56% |
| streptavidin (5) | 23,178 | 14,901 | 3,692 | 4,090 | 398 | 10.78% |
|
|
||||||
| total (57) | 282,989 | 184,118 | 39,159 | 44,419 | 5,260 | 13.43% |
Raw number of molecules obtained using each method.
Filtered number of molecules using D3N-drugc target ranges.
Relative increase in filtered molecules (D3N-drugc vs D3N-loose) protocols (# molecules and %).
Figure 12.

Added value for on-the-fly pruning versus hard-cut filtering. Panels compare D3N-loose (gray, 39,159 molecules) and D3N-drugc (red, 44,419 molecules) from ensembles filtered to remove molecules with descriptor values outside the D3N-drugc target ranges.
As an additional point of comparison, for how most users would likely employ de novo design in a practical setting, we compared the results obtained using the “on-the-fly pruning” approach which yields smooth-tailed distributions (D3N-drugc) versus a brute-force “build-all-then-filter” approach which yields hard-cut distributions (D3N-loose filtered). Figure S2 visually highlights the differences between the two approaches in terms of cheminformatic scores across the eight protein families. From an energetic standpoint, Figure 13 plots DOCK6 grid scores (non-bonded protein-ligand VDW + ES energy) along with the number of top-scoring molecules for the range −50 kcal/mol and below (left of vertical black dotted line). As expected, across all protein families (57 systems), the aggressive pruning strategy in conjunction with Metropolis-like smoothing (a desirable attribute of the D3N-drugc method) yields significant enrichment compared to the brute-force filtered approach (Figure 13, bottom right plot 23,308 red vs 1,556 gray molecules). Across each individual protein family, the enrichment was varied. For example, growth in acetylcholinesterase yielded 2,786 additional molecules (2,999 red vs 213 gray) while growth in cyclooxygenase yielded a smaller increase (655 red vs 49 gray). For HIVPR, the increase was particularly large (10,700 red vs 427 gray).
Figure 13.

DOCK6 energy scores from molecules in the D3N-drugc ensemble (red line) versus molecules in the D3N-loose ensemble filtered afterward by the D3N-drugc target ranges (gray area) by protein family. Energies in kcal/mol.
The analysis above emphasizes the key differences likely to occur as a result of using either modeling approach out of the box. From an academic standpoint however, it is noteworthy that there is also enrichment when the underlying smoothed-tail D3N-drugc results are hard-cut filtered and only the molecules with DOCK6 grid scores < −50 kcal/mol are retained. Although, in practice, users would have no need to filter out the D3N-drugc outcomes since by design the method already biases the descriptor distributions thus the analysis here is largely theoretical. Nevertheless, with both datasets hard-filtered, as shown in Figure 14, use of D3N-drugc yields enrichment in the number of top scoring molecules versus D3N-loose (red vs gray bars) in 7 out of 8 cases. While the numerical improvements (indicated above each red bar) are not as large as the analysis shown in Figure 13, they do establish a desirable outcome. For example, many research groups (especially smaller academic labs) may only have resources to synthesize and/or purchase a limited number of molecules for biological testing for any given lead-discovery project. Thus, any enrichment in the number of “quality” molecules, in terms of their cheminformatics properties and protein-ligand scores, is likely to be beneficial, especially for those in the top-ranked range (DOCK6 scores −50 kcal/mol and below). While additional testing is necessary to determine if these are general trends, the results in this section provides strong evidence that on-the-fly layer-by-layer pruning adds quantitative value relative to a build-all and filter approach.
Figure 14.

Number of top-scoring molecules from D3N-drugc filtered and D3N-loose filtered protocols for the DOCK6 grid score range −50 kcal/mol and below arranged by protein family. Values above each red bar indicate increases arising from use of the D3N-drugc filtered versus D3N-loose filtered protocol.
Apparent influence of the protein environment on druglikeness.
Interestingly, a comparison between molecules constructed using the D3N-loose protocol in protein binding sites (D3N-loose_protein=yes) and those constructed in the absence of protein (D3N-loose_protein=no) brings to light an implicit bias towards the construction of compounds with more favorable QED scores prior to use of any RDKit descriptors. As shown in Figure 15, D3N-loose_protein=no simulations generate compounds with QED scores closer to 0 (less drug like) which show a strong well-defined peak at about 0.25 (gray shade). Conversely, the same D3N-loose protocols, but executed in protein binding sites (D3N-loose_protein=yes, green), yield a right-shifted QED distribution with population peak at about 0.7 (more drug like). Further, this D3N-loose_protein=yes profile for QED shows remarkable agreement with the distribution generated from approved drugs and active pharmaceutical agents in the DrugC dataset (Figure 15, red). This interesting result suggests that the protein environment alone inherently biases de novo growth towards the generation of more drug-like molecules. It is likely that this observation is multi-faceted, but attributable in part to the physics-based scoring functions used during growth for ranking and pruning. In the absence of protein, (simple-build protocol) the primary energy function employed is a simple non-bonded intramolecular energy of the ligand (VDW repulsive term) which primarily enforces linear conformations. In contrast, in protein environments, the primary energy function is the standard DOCK grid score comprised of the pairwise sum of all non-bonded intermolecular interactions between the protein and ligand (VDW plus ES energies, see Methods).
Figure 15.

Comparison of QED scores, and the eight underlying descriptors that make up QED, derived from ensembles generated in the absence of a proton (gray, D3N-loose_protein=no) and in protein binding sites (green, D3N-loose_protein=yes). For comparison, distributions from molecules in the DrugC dataset are also plotted (red, DrugC). MW in g/mol, TPSA in angstroms squared.
An examination of the underlying terms that make up the QED score reveals that the D3N-loose_protein=yes simulations (green) yield smaller and more compact molecules, compared to D3N-loose_protein=no simulations (gray), as highlighted by the shifts in MW and TPSA subplots (Figure 15 green vs gray). Here, D3N-loose_protein=no yield molecules with MW that shift towards the user-defined upper limit, in this case, 550 g/mol. In contrast, D3N-loose_protein=yes, yields a MW distribution peaking around ~250). Since the terms are not being explicitly driven it is likely that the trend towards smaller MW is a result of binding sites having finite volume. The other descriptors in Figure 15, with the exception of LogP and #Alerts, would also be expected to be correlated with molecular size, in particular, smaller numbers of ligand #Rotatable bonds, smaller TPSA, and fewer aromatic rings. As pointed out by a reviewer, our observations also likely reflect the choice of proteins employed in the study, for which all are drug targets with the exception of streptavidin. And for binding pockets with different character (i.e. larger or more polar), it can be speculated that the ligand properties would change accordingly. In any event, the data in Figure 15 suggests that simulations in proteins (green), of similar character as studied here, will have an inherent advantage in terms of generating more “drug-like” molecules using metrics such as QED with property distributions similar to compounds in the DrugC dataset (red). Use of additional biasing, via the new DOCK6/RDKit interface, in some sense, makes an already reasonable situation even better. In contrast, if using the current DOCK6 protocols to design compounds in the absence of protein, it would be recommended to apply D3N-drugc ranges in Table 3, or other equivalent user-defined ranges, as these boundary conditions help bias construction towards drug-like space.
D3N protocols can yield “pinpoint” descriptor ranges.
The results in Figure 10 confirm that DOCK_D3N can be used to grow molecules from scratch in a protein binding site that fall within a user-defined set of ranges for descriptors. In this section, we explore the ability of the algorithm to generate molecules with enhanced protein interactions while simultaneously matching descriptor values of a given reference ligand (termed D3N-pinpoint protocol). For these experiments, six clinically-relevant protein-ligand systems from our SB201223,39 test set were selected: (1) SB203580 with MAP kinase (pdb 1A9U),53 (2) SU2 with FGR1 kinase domain (pdb 1AGW),54 (3) flurbiprofen with COX-1 (pdb 1EQH),55 (4) simvastatin with HMG-CoA reductase (pdb 1HW9),56 (5) efavirenz with HIV reverse transcriptase (pdb 1IKW),57 and (6) oseltamivir with neuraminidase (pdb 3CL0).58 Table 6 shows descriptor values for each reference ligand (computed using DOCK6/RDKit) along with the D3N-pinpoint target ranges derived from each reference for the five D3N descriptors being driven.
Table 6.
Descriptor values for reference ligands and derived target ranges for D3N-pinpoint simulations.
| pdb code | QED | SynthA | TPSA | LogP | #Stereo | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| ref liganda | D3N-pinpointb | ref ligand | D3N-pinpoint | ref ligand | D3N-pinpoint | ref ligand | D3N-pinpoint | ref ligand | D3N-pinpoint | |
| 1A9U | 0.56 | 0.46 | 2.84 | 3.84 | 58.6 | 48 to 68 | 4.68 | 3.68 to 5.68 | 1 | 2 |
| 1AGW | 0.69 | 0.59 | 2.34 | 3.34 | 52.7 | 42 to 62 | 2.46 | 1.46 to 3.46 | 0 | 1 |
| 1EQH | 0.83 | 0.72 | 2.65 | 3.65 | 40.1 | 30 to 50 | 2.35 | 1.34 to 3.34 | 1 | 2 |
| 1HW9 | 0.51 | 0.41 | 4.89 | 5.88 | 106.9 | 96 to 116 | 2.77 | 1.77 to 3.77 | 7 | 8 |
| 1IKW | 0.73 | 0.63 | 3.57 | 4.56 | 38.3 | 28 to 48 | 4.07 | 3.07 to 5.07 | 1 | 2 |
| 3CL0 | 0.64 | 0.53 | 4.82 | 5.81 | 106.1 | 96 to 116 | −1.24 | −2.20 to −0.20 | 3 | 4 |
Descriptor values for reference ligands.
Target ranges for D3N-pinpoint calculations (std dev: QED = 0.05, SynthA = 0.10, TPSA = 5.0, LogP = 0.10). TPSA values in angstroms squared.
Preliminary tests showed that the very extreme pinpoint ranges in Table 6 led to excessive pruning. To bolster sampling under these conditions, we increased the values for three key de novo design parameters: dn_num_random_picks (20 to 50), dn_max_root_size (25 to 50), and dn_max_layer_size (25 to 50). We also increased the number of anchors used to seed ligand growth (15 to 380). As noted in methods, the pinpoint simulations also included footprint similarity (FPS) terms20,36 in the primary scoring function which helps bias growth towards per-residue VDW and ES patterns made by the reference ligand. The overall objective was to generate topologically different molecules but with characteristics similar to the reference. For each system, the ensembles resulting from different anchors were pooled together, rank ordered, and the top 500 molecules were retained for analysis.
Figure 16 plots D3N-pinpoint results with subplots ordered based on the scores for the six reference ligands (ref ligand rank in bold font, plots arranged from low to high). Readers should note that the order of systems for the three descriptors are different. For example, QED values for the six references go from 0.51 (1HW9) to 0.83 (1EQH), TPSA goes from 38.3 (1IKW) to 106.9 (1HW9), and LogP goes from −1.24 (3CL0) to 4.68 (1A9U). Notably, the associated distributions from the D3N-pinpoint simulations appear to be well-correlated in terms of the reference ligand rank trends. For example, the QED score for the reference ligand in 1HW9 (0.51) is the lowest among the six systems examined and the resultant QED profile from D3N-point simulations in 1HW9 is shifted farthest left. Conversely, the reference ligand in 1EQH has the highest QED score (0.83) and the D3N-pinpoint distribution is shifted furthest right. The recognizable left to right progression, while traveling down each row, for the three descriptors plotted, provides evidence the D3N algorithm can be used to tune molecular outcomes about a desired descriptor value.
Figure 16.

Descriptor distributions for the top 500 molecules, constructed using D3N-pinpoint protocols (Table 6), with subplots arranged in increasing order based on the QED, TPSA, or LogP values for the six reference ligands.
D3N searching yields enhanced interactions.
An examination of top-scoring molecule from the D3N-pinpoint simulations shows there is significant chemical searching about each binding site which can be visualized by plotting H-bond patterns. Figure 17 shows specific H-bond interactions (discussions employ pdb numbering) made by the six reference ligands (top panels, green ligand, magenta labeled residues) with those made by the 500 top-scoring molecules constructed during each D3N simulation (bottom panels, magenta and orange labeled residues). To emphasize trends, molecules grown using D3N are hidden and only a subset of binding site residues are shown. Reassuringly, in each case, newly constructed molecules recapitulate key ES interactions made by each reference (magenta labeled residues) as well explore additional regions in the binding site (orange labeled residues). For example, D3N simulations in Map kinase (Figure 17a) yield ligand ensembles with highly populated H-bonds at residues Lys53 and Met109 (bottom panel, magenta residues) which correspond to the two H-bonds made by the reference ligand (top panel, magenta residues). And, the algorithm explores additional H-bonding which is observed at positions Ala51, Leu104, Gly110, and Asp168 (bottom panel, orange residues). Likewise, for the FGR1 kinase domain (Figure 17b), the D3N ensembles yield significant H-bond populations at Gly562 and Ala564 (bottom panel, magenta residues), corresponding to those made by the reference ligand (top panel, magenta residues), and there are additional interaction patterns seen with Lys514, Asn568, and Tyr563 (bottom panel, orange residues). An examination of the remaining four protein systems in Figure 17 (c-f) show similar trends.
Figure 17.

H-bond patterns (dashed magenta lines) made by six reference ligands (top panels, ref ligand pose in green) and top-scoring ensembles (N=500 molecules) from D3N-pinpoint simulations (bottom panels, D3N molecules hidden for clarity) for (a) SB203580 with Map kinase (1A9U), SU2 with FGR1 kinase domain (1AGW), flurbiprofen with COX-1 (1EQH), simvastatin HMG-CoA reductase (1HW9), efavirenz with HIV-1RT (1IKW), and oseltamivir with neuraminidase (3CL0). H-bonds calculated using the Chimera59 program with default settings. Select binding site residues shown in gray.
In terms of specific molecules, Figure 18 compares the pose of each reference ligand (green) with the top scoring pose from each D3N simulation (orange) along with their respective MGE energies (VDW + ES terms) and RDKit descriptor scores for QED, SynthA, TPSA, and LogP. As expected, the descriptor values from the D3N-pinpoint simulations mirror their respective references (Figure 18 tables, reference ligand versus D3N top score). As an example, for 1A9U, both TPSA scores (58.64, 72.28) and both LogP scores (4.68, 5.53) are similar in terms of numerical value. And for 3CL0, the D3N scores shift accordingly to mimic the larger TPSA (106.10, 121.70) and negative LogP (−1.24, −0.18) values of the reference. The molecules also show reasonable 3D overlap in the binding site which occurs in part because the MGS+FPS scoring function helps keep growth from expanding too far outside of the binding pocket. Descriptor comparisons for the four other systems in Figure 18 follow similar trends. Of particular interest, in four out of six cases (1A9U, 1AGW, 1IKW, 3CL0), the top scoring D3N molecules in Figure 18 yield a more favorable MGE score relative to its reference. This favorable outcome suggests that the D3N pinpoint protocol will be useful to explore generation of new compounds, with enhanced binding site interactions, while conforming to an underling descriptor space defined by a known compound or reference.
Figure 18.

Comparison between the top-scoring D3N-pinpoint pose (orange), and its respective reference ligand pose (green), along with MGE energy, QED, SynthA, TPSA, and LogP scores. Protein residues hidden for clarity. MGE in kcal/mol. TPSA in angstroms squared.
Conclusions.
In conclusion, this work presents development and testing of a new de novo design (DN) approach for the program DOCK6 termed Descriptor Driven De Novo (D3N) which aims to bias the construction of new small organic molecules to conform to a set of user-defined properties. The new D3N method makes use of descriptors computed through integration of DOCK6 with the open-source cheminformatics package RDKit. At each stage (layer) of ligand growth, if values for given descriptor (or descriptors) fall outside of the target range(s) defined by the user, the molecule is likely pruned (Figure 1). Layer-by-layer growth requires fragment libraries, which for the present work, were derived from deconstruction of 13M drug-like molecules (Figure 2). D3N pruning makes use of a Metropolis-like scheme, permitting some molecules at the descriptor boundaries to propagate, which yields smooth-tailed distributions (Figure 3). Pruning is also executed in coordination with existing routines which control, for example, molecular and conformational diversity, size, formal charge, and interaction energy. The DOCK6/RDKit method can be used to bias growth based on a single descriptor at a time, a single descriptor that includes multiple underlying terms in the function, or multiple descriptors simultaneously. The current implementation allows for 13 RDKit descriptors to be computed (see Introduction) of which 7 can be used for D3N growth. The simulations discussed in this work employed up to 5 descriptors simultaneously (QED, SynthA, TPSA, LogP, #Stereo). The new DOCK6/RDKit implementation yielded results essentially identical to the standard Python/RDKit distribution, for 13 million molecules and 9 descriptors, thereby confirming the integrity of the RDKit integration (Figure 5). The validation process also confirmed the reliability of MOL2 to SMILES conversions (Figure 6). The DOCK6/RDKit implementation can also be used to process large ligand libraries (via the database filter/utilities class) to help prioritize compounds for standard virtual screening, either before or after docking has occurred. As an example, users can easily explore different rank ordering options using DOCK6 MOL2 files containing descriptors values and other scoring terms in conjunction with the UCSF Chimera program (ViewDock utility).
In comprehensive testing, five different D3N protocols were evaluated, including those that specified loose ranges with little to no pruning (D3N-loose), ranges based on approved small molecule drugs and active pharmaceutical agents (D3N-drugc), ranges designed to laterally shift D3N-drugc distributions left or right (D3N-lateral), ranges that were narrower than the D3N-drugc targets (D3N-narrow), and ranges designed to give tight pinpoint distributions (D3N-pinpoint). Notably, in all cases, use of these different protocols, relative to using D3N-loose as a control, led to descriptor populations that shift left or right, or are narrower, which indicates that the DOCK6/RDKit D3N infrastructure is working as intended. Particularly striking changes were observed using the D3N-lateral protocol in the absence of protein (Figures 7 red vs gray), the D3N-narrow protocol in 57 protein systems (Figure 10 purple vs gray), and the D3N-pinpoint protocol in 6 protein binding sites (Figure 16, Table 6). Layer-by-layer growth experiments confirmed that the D3N protocol can be initiated at any point during a DOCK_DN simulation, and descriptor ranges approach their targets more completely when the algorithm is applied in earlier layers (Figure 9). Our studies also highlight that for functions such as QED, comprised of multiple descriptors, driving the primary function alone will show concomitant changes in the underlying descriptors themselves (Figure 9). Outcomes for LogP calculations were also observed to be influenced by the underlying composition of the fragment libraries used during ligand growth (Figure 7).
For the D3N-drugc simulations in 57 proteins, relative to the D3N-loose control, a comparison of results hard-filtered to the same cutoffs showed that there was added value in terms of an increase in the number of designed molecules (13.4%) with more favorable QED, SynthA, TPSA, and LogP scores (Figure 12 red vs gray area, Table 5). By protein family, the increases ranged from 4.87% to 25.56% (Table 5). And for the top-scoring range, grid scores of −50 kcal/mol and below, the D3N-drugc filtered protocol yielded more molecules than D3N-loose filtered for 7 out 8 protein families (Figure 14). A particularly interesting observation was the apparent influence on increased druglikeness (QED) when growth was initiated in binding sites, compared to the absence of protein, before additional D3N biasing (Figure 15). An examination of the underlying QED terms showed that molecules constructed in binding sites were smaller which is likely due to differences in scoring function (internal energy only versus non-bonded protein interactions) and the sites being of finite volume. Simulations using the D3N-pinpoint protocol, for 6 clinically relevant drug targets, showed that the algorithm was effective at generating descriptor distributions which tracked the descriptor trends made by the cognate reference ligand (Figure 16). An examination of H-bond patterns, for the 500 top-scoring molecules in each of the 6 sites, showed high density with residues known to be engaged by the reference, and at other sites, which indicates robust chemical searching and sampling (Figure 17). And in 4 out of 6 cases, the top-scoring D3N molecule yielded a DOCK energy score more favorable than the reference while retaining similar descriptor values (Figure 18).
In summary, the new D3N approach is an important building block of our long-term strategy to develop an extensive de novo design platform for which the whole is greater than the sum of the individual parts. The comprehensive experiments outlined in this manuscript indicate that the DOCK_D3N algorithm will aggressively prune molecules when descriptors fall outside of the target ranges leading to descriptor populations enriched for specific properties. Planned future work includes implementation of additional RDKit descriptors and adopting the interface for use with the DOCK6 genetic algorithm (GA) recently reported by Prentis et al.26 As with all our prior development efforts, the D3N method will be included in a forthcoming public release of DOCK for use by the community. We hypothesize that the GA interface may show enhanced convergence because the limitation that molecules must be correctly built to “spec” in only nine layers of de novo growth will be lifted.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grant R35GM126906 (to R.C.R) with student support from T32GM136572 (to S.P). The authors would like to thank past and present Rizzo Lab members for computational assistance and discussion. In particular, Dr. Lauren Prentis and John Bickel for algorithm discussion, Dr. Trent Balius for implementing the DOCK6 simple-build scoring function, and Christopher Corbo for organizing the DrugCentral dataset. Additional thanks are given to the Stony Brook University Office of the Vice President for Research, Stony Brook Research Computing and Cyberinfrastructure, and the Institute for Advanced Computational Science at Stony Brook University for access to the high-performance LIred and SeaWulf computing systems, the latter of which was made possible by a National Science Foundation grant (#1531492).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.3c01031
Procedures used to determine default descriptor ranges and comparisons between DrugCentral and ZINC databases. Comparison of results using D3N-pinpoint and D3N-loose protocols (PDF).
Complete contact information is available at: https://pubs.acs.org/doi/10.1021/acs.jcim.3c01031
The authors declare no competing financial interest.
Data And Software Availability
The RDKit interface for DOCK6 described in this work will be made available for use by the community to coincide with publication of the manuscript, or shortly thereafter. Until the official release date, interested users with a valid DOCK6 license can obtain the software directly from the corresponding author. DOCK6 is free for academics, including all source code, and is available at https://dock.compbio.ucsf.edu/DOCK_6/index.htm. RDKit is open-source and available at https://www.rdkit.org. Compilation will require the 2019.09.01 release of RDKit (www.rdkit.org), Boost 1.71.0 (www.boost.org), Eigen 3.3.9 (www.eigen.tuxfamily.org), and Anaconda3 (www.anaconda.com). Protein simulations employed systems in the SB2012 protein data set available at ringo.ams.stonybrook.edu/index.php/Rizzo_Lab_Downloads. Data analysis and plotting was performed with pandas v1.1.4 (www.pandas.pydata.org) and matplotlib v3.6.0 (www.matplotlib.org) in conjunction with python v3.9 (www.python.org). The program UCSF Chimera (www.cgl.ucsf.edu/chimera) was also used for data analysis and visualization.
References
- (1).Shoichet BK Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sterling T; Irwin JJ ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model 2015, 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Irwin JJ; Sterling T; Mysinger MM; Bolstad ES; Coleman RG ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Irwin JJ; Shoichet BK ZINC - A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Moon JB; Howe WJ Computer design of bioactive molecules: a method for receptor-based de novo ligand design. Proteins: Struct., Funct., Genet 1991, 11, 314–328. [DOI] [PubMed] [Google Scholar]
- (6).Pellegrini E; Field MJ Development and testing of a de novo drug-design algorithm. J. Comput.-Aided Mol. Des 2004, 17, 621–641. [DOI] [PubMed] [Google Scholar]
- (7).Talamas FX; Ao-Ieong G; Brameld KA; Chin E; de VJ; Dunn JP; Ghate M; Giannetti AM; Harris SF; Labadie SS; Leveque V; Li J; Lui AST; McCaleb KL; Najera I; Schoenfeld RC; Wang B; Wong A. De Novo Fragment Design: A Medicinal Chemistry Approach to Fragment-Based Lead Generation. J. Med. Chem 2013, 56, 3115–3119. [DOI] [PubMed] [Google Scholar]
- (8).Schneider G. De novo design - hop(p)ing against hope. Drug Discovery Today: Technol. 2013, 10, e453–e460. [DOI] [PubMed] [Google Scholar]
- (9).Hartenfeller M; Schneider G. Enabling future drug discovery by de novo design. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2011, 1, 742–759. [Google Scholar]
- (10).Loving K; Alberts I; Sherman W. Computational approaches for fragment-based and de novo design. Curr. Top. Med. Chem 2010, 10, 14–32. [DOI] [PubMed] [Google Scholar]
- (11).Schneider G; Fechner U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discovery 2005, 4, 649–663. [DOI] [PubMed] [Google Scholar]
- (12).Schneider P; Schneider G. De Novo Design at the Edge of Chaos. J. Med. Chem 2016, 59, 4077–4086. [DOI] [PubMed] [Google Scholar]
- (13).Xie W; Wang F; Li Y; Lai L; Pei J. Advances and Challenges in De Novo Drug Design Using Three-Dimensional Deep Generative Models. J. Chem. Inf. Model 2022, 62, 2269–2279. [DOI] [PubMed] [Google Scholar]
- (14).Mouchlis VD; Afantitis A; Serra A; Fratello M; Papadiamantis AG; Aidinis V; Lynch I; Greco D; Melagraki G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci 2021, 22, 1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Domenico A; Nicola G; Daniela T; Fulvio C; Nicola A; Orazio N. De Novo Drug Design of Targeted Chemical Libraries Based on Artificial Intelligence and Pair-Based Multiobjective Optimization. J. Chem. Inf. Model 2020, 60, 4582–4593. [DOI] [PubMed] [Google Scholar]
- (16).Merk D; Grisoni F; Friedrich L; Schneider G. Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators. Commun. Chem 2018, 1, 68. [Google Scholar]
- (17).Merk D; Friedrich L; Grisoni F; Schneider G. De Novo Design of Bioactive Small Molecules by Artificial Intelligence. Mol. Inf 2018, 37, 1700153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bjerrum EJ; Sattarov B. Improving Chemical Autoencoder Latent Space and Molecular De Novo Generation Diversity with Heteroencoders. Biomolecules 2018, 8, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cheron N; Jasty N; Shakhnovich EI OpenGrowth: An Automated and Rational Algorithm for Finding New Protein Ligands. J. Med. Chem 2016, 59, 4171–4188. [DOI] [PubMed] [Google Scholar]
- (20).Allen WJ; Fochtman BC; Balius TE; Rizzo RC Customizable de novo design strategies for DOCK: Application to HIVgp41 and other therapeutic targets. J. Comput. Chem 2017, 38, 2641–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ewing TJA; Kuntz ID Critical evaluation of search algorithms for automated molecular docking and database screening. J. Comput. Chem 1997, 18, 1175–1189. [Google Scholar]
- (22).RDKit: Open-source cheminformatics. https://www.rdkit.org;
- (23).Allen WJ; Balius TE; Mukherjee S; Brozell SR; Moustakas DT; Lang PT; Case DA; Kuntz ID; Rizzo RC DOCK 6: Impact of new features and current docking performance. J. Comput. Chem 2015, 36, 1132–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brozell SR; Mukherjee S; Balius TE; Roe DR; Case DA; Rizzo RC Evaluation of DOCK 6 as a pose generation and database enrichment tool. J. Comput-Aided Mol. Des 2012, 26, 749–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lang PT; Brozell SR; Mukherjee S; Pettersen EF; Meng EC; Thomas V; Rizzo RC; Case DA; James TL; Kuntz ID DOCK 6: combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Prentis LE; Singleton CD; Bickel JD; Allen WJ; Rizzo RC A molecular evolution algorithm for ligand design in DOCK. J. Comput. Chem 2022, 43, 1942–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bickerton GR; Paolini GV; Besnard J; Muresan S; Hopkins AL Quantifying the chemical beauty of drugs. Nat. Chem 2012, 4, 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ertl P; Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminf 2009, 1, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ertl P; Rohde B; Selzer P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem 2000, 43, 3714–3717. [DOI] [PubMed] [Google Scholar]
- (30).Wildman SA; Crippen GM Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci 1999, 39, 868–873. [Google Scholar]
- (31).Delaney JS ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci 2004, 44, 1000–1005. [DOI] [PubMed] [Google Scholar]
- (32).Capuzzi SJ; Muratov EN; Tropsha A. Phantom PAINS: Problems with the Utility of Alerts for Pan-Assay INterference CompoundS. J. Chem. Inf. Model 2017, 57, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Baell JB Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod 2016, 79, 616–628. [DOI] [PubMed] [Google Scholar]
- (34).Baell JB; Holloway GA New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- (35).Durant JL; Leland BA; Henry DR; Nourse JG Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci 2002, 42, 1273–1280. [DOI] [PubMed] [Google Scholar]
- (36).Balius TE; Allen WJ; Mukherjee S; Rizzo RC Grid-based molecular footprint comparison method for docking and de novo design: Application to HIVgp41. J. Comput. Chem 2013, 34, 1226–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Balius TE; Mukherjee S; Rizzo RC Implementation and evaluation of a docking-rescoring method using molecular footprint comparisons. J. Comput. Chem 2011, 32, 2273–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gasteiger J; Marsili M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar]
- (39).Mukherjee S; Balius TE; Rizzo RC Docking Validation Resources: Protein Family and Ligand Flexibility Experiments. J. Chem. Inf. Model 2010, 50, 1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jakalian A; Bush BL; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic Charges. AM1-BCC model: I. Method. J. Comput. Chem 2000, 21, 132–146. [DOI] [PubMed] [Google Scholar]
- (42).Jakalian A; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem 2002, 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- (43).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (44).Wang J; Wang W; Kollman PA; Case DA Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- (45).Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ The Amber biomolecular simulation programs. J. Comput. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Case DA; Cerutti DS; Cheatham TE 3rd; Darden TA; Duke RE; Giese TJ; Gohlke H; Goetz AW; Greene D; Homeyer N; Izadi S; Kovalenko A; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein D; Merz KM; Monard G; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Simmerling CL; Botello-Smith WM; Swails J; Walker RC; Wang J; Wolf RM; Wu X; Xiao L; York DM; Kollman PA AMBER 16. University of California, San Francisco 2016. [Google Scholar]
- (48).DMS; UCSF Computer Graphics Laboratory; San Francisco, CA [Google Scholar]
- (49).DesJarlais RL; Sheridan RP; Seibel GL; Dixon JS; Kuntz ID; Venkataraghavan R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. J. Med. Chem 1988, 31, 722–729. [DOI] [PubMed] [Google Scholar]
- (50).Meng EC; Shoichet BK; Kuntz ID Automated docking with grid-based energy evaluation. J. Comput. Chem 1992, 13, 505–524. [Google Scholar]
- (51).Ursu O; Holmes J; Knockel J; Bologa CG; Yang JJ; Mathias SL; Nelson SJ; Oprea TI DrugCentral: online drug compendium. Nucleic Acids Res. 2017, 45, D932–d939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Avram S; Bologa CG; Holmes J; Bocci G; Wilson TB; Nguyen D-T; Curpan R; Halip L; Bora A; Yang JJ; Knockel J; Sirimulla S; Ursu O; Oprea TI DrugCentral 2021 supports drug discovery and repositioning. Nucleic Acids Res. 2021, 49, D1160–D1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wang Z; Canagarajah BJ; Boehm JC; Kassisa S; Cobb MH; Young PR; Abdel-Meguid S; Adams JL; Goldsmith EJ Structural basis of inhibitor selectivity in MAP kinases. Structure 1998, 6, 1117–1128. [DOI] [PubMed] [Google Scholar]
- (54).Mohammadi M; McMahon G; Sun L; Tang C; Hirth P; Yeh BK; Hubbard SR; Schlessinger J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997, 276, 955–960. [DOI] [PubMed] [Google Scholar]
- (55).Selinsky BS; Gupta K; Sharkey CT; Loll PJ Structural analysis of NSAID binding by prostaglandin H2 synthase: time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry 2001, 40, 5172–5180. [DOI] [PubMed] [Google Scholar]
- (56).Istvan ES; Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [DOI] [PubMed] [Google Scholar]
- (57).Lindberg J; Sigurdsson S; Lowgren S; Andersson HO; Sahlberg C; Noreen R; Fridborg K; Zhang H; Unge T. Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem 2002, 269, 1670–1677. [DOI] [PubMed] [Google Scholar]
- (58).Collins PJ; Haire LF; Lin YP; Liu J; Russell RJ; Walker PA; Skehel JJ; Martin SR; Hay AJ; Gamblin SJ Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature 2008, 453, 1258–1261. [DOI] [PubMed] [Google Scholar]
- (59).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RDKit interface for DOCK6 described in this work will be made available for use by the community to coincide with publication of the manuscript, or shortly thereafter. Until the official release date, interested users with a valid DOCK6 license can obtain the software directly from the corresponding author. DOCK6 is free for academics, including all source code, and is available at https://dock.compbio.ucsf.edu/DOCK_6/index.htm. RDKit is open-source and available at https://www.rdkit.org. Compilation will require the 2019.09.01 release of RDKit (www.rdkit.org), Boost 1.71.0 (www.boost.org), Eigen 3.3.9 (www.eigen.tuxfamily.org), and Anaconda3 (www.anaconda.com). Protein simulations employed systems in the SB2012 protein data set available at ringo.ams.stonybrook.edu/index.php/Rizzo_Lab_Downloads. Data analysis and plotting was performed with pandas v1.1.4 (www.pandas.pydata.org) and matplotlib v3.6.0 (www.matplotlib.org) in conjunction with python v3.9 (www.python.org). The program UCSF Chimera (www.cgl.ucsf.edu/chimera) was also used for data analysis and visualization.