
Abstract
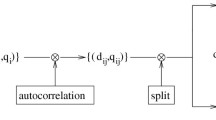
Correlation vector methods were tested for their usefulness in ligand-based virtual screening. Three molecular descriptors – two based on potential pharmacophore points and one on partial atom charges – and three similarity measures – the Manhattan distance, the Euclidian distance and the Tanimoto coefficient – were compared. The alignment-free descriptors seem to be particularly applicable when a course-grain filtering of data sets is required in combination with a high execution speed. Significant enrichment of actives was obtained by retrospective analysis. The cumulative percentages for all three descriptors allow for the retrieval of up to 78% of the active molecules in the first five percent of the reference database. Different descriptors retrieved only weakly overlapping sets of active molecules among the top-ranking compounds. If a single similarity index is to be used, the Manhattan distance seems to be particularly applicable. Generally, none of the three different descriptors tested in this study clearly outperformed the others. The suitability of a descriptor critically depends on the ligand-receptor interaction under investigation. For ligand-based similarity searching it is recommended to exploit several descriptors in parallel.
Similar content being viewed by others


References
Barnard, J.M., Downs, G.M. and Willett, P. Descriptor-Based Similarity Measures for Screening Chemical Databases, in Böhm, H.J., and Schneider, G. (Eds.) Virtual Screening of bioactive Molecules, Wiley-VCH 2000, Weinheim, New York, 59-80.
Schneider, G. and Nettekoven, M., J. Comb. Chem., 5 (2003) 233.
Schuffenhauer A., Floersheim P., Acklin P. and Jacoby E., J. Chem. Inf. Comput. Sci., 43 (2003) 391.
Stahl, M., Rarey, M. and Klebe, G., Screening of drug databases, in Lengauer, T. (Ed.) Bioinformatics: From Genomes to Drugs Vol. 2, Wiley-VCH 2001, Weinheim, New York, 137-170.
Broto, P., Moreau, G. and Vandyke, C., Eur. J. Med. Chem., 19 (1984) 66.
Bauknecht, H., Zell, A., Bayer, H., Levi, P., Wagener, M., Sadowski, J. and Gasteiger, J., J. Chem. Inf. Comput. Sci., 36 (1996) 1205.
Anzali, S., Barnickel, G., Krug, M., Sadowski, J., Wagener, M., Gasteiger, J. and Polanski, J., J. Comput. Aided Mol. Des., 10 (1996) 521.
Zupan, J. and Gasteiger, J., Neural Networks in Chemistry and Drug Design, Wiley-VCH 1999, Weinheim, New York.
Schneider, G., Neidhart, W., Giller, T. and Schmid, G., Angew. Chem. Int. Ed., 38 (1999) 2894.
Schneider, G., Chomienne-Clement, O., Hilfiger, L., Kirsch, S., Böhm, H.-J., Schneider, P. and Neidhart, W., Angew. Chemie Int. Ed., 39 (2000) 4130.
Schneider, P. and Schneider, G., QSAR Comb. Sci., 22 (2003) in press.
Carhart, R.E., Smith, D.H. and Venkataraghavan, R., J. Chem. Inf. Comput. Sci., 25 (1985) 64.
MOE, Molecular Operating Environment. Distributor: Chemical Computing Group, 1010 Sherbrooke St. West, #910, Montreal, Canada H3A.
Bush, B.L. and Sheridan, R.P., J. Chem. Inf. Comput. Sci., 33 (1993) 756.
CLIFF, CORINA, PETRA. Distributor: Molecular Networks GmbH, Computerchemie, Nägelsbachstrasse 25, D-91052 Erlangen, Germany, http://www.mol-net.de/.
Gasteiger, J., Rudolph, C. and Sadowski, J., Tetrahedron Comp. Method., 3 (1990) 537.
Willett, P, Barnard, J.M. and Downs, G., J. Chem. Inf. Comput. Sci., 38 (1998) 983.
Xu, H. and Agrafiotis, D.K., Curr. Top. Med. Chem., 2 (2002) 1305.
Corman, T.H., Leiserson, C.E., Rivest, R.L. and Stein, C., Introduction to Algorithms, 2nd Edition, MIT Press, Cambridge, 2001.
Brown, R.D. and Martin, Y.C., J. Chem. Inf. Comput. Sci., 37 (1997) 1.
Brown, R.D. and Martin, Y.C., J. Chem. Inf. Comput. Sci., 36 (1996) 572.
Sadowski, J., J. Comput. Aided Mol. Des., 11 (1997) 53.
Xu, H., Izrailev, S. and Agrafiotis, D., J. Chem. Inf. Comput. Sci., 43 (2003) 1186.
Holliday, J.D., Salim, N., Whittle, M. and Willett, P., J. Chem. Inf. Comput. Sci., 43 (2003) 819.
Chen, X. and Reynolds, C.H., J. Chem. Inf. Comput. Sci., 42 (2002) 1407.
Schneider, G., Curr. Med. Chem., 9 (2002) 2095.
Tropsha, A. and Zheng, W., Comb. Chem. High Throughput Screen., 5 (2002) 111.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Fechner, U., Franke, L., Renner, S. et al. Comparison of correlation vector methods for ligand-based similarity searching. J Comput Aided Mol Des 17, 687–698 (2003). https://doi.org/10.1023/B:JCAM.0000017375.61558.ad
Issue Date:
DOI: https://doi.org/10.1023/B:JCAM.0000017375.61558.ad