

Abstract
Alternative polyadenylation (APA) in breast tumor samples results in the removal/addition of cis-regulatory elements such as microRNA (miRNA) target sites in the 3′-untranslated region (3′-UTRs) of genes. Although previous computational APA studies focused on a subset of genes strongly affected by APA (APA genes), we identify miRNAs of which widespread APA events collectively increase or decrease the number of target sites [probabilistic inference of microRNA target site modification through APA (PRIMATA-APA)]. Using PRIMATA-APA on the cancer genome atlas (TCGA) breast cancer data, we found that the global APA events change the number of the target sites of particular microRNAs [target sites modified miRNA (tamoMiRNA)] enriched for cancer development and treatments. We also found that when knockdown (KD) of NUDT21 in HeLa cells induces a different set of widespread 3′-UTR shortening than TCGA breast cancer data, it changes the target sites of the common tamoMiRNAs. Since the NUDT21 KD experiment previously demonstrated the tumorigenic role of APA events in a miRNA dependent fashion, this result suggests that the APA-initiated tumorigenesis is attributable to the miRNA target site changes, not the APA events themselves. Further, we found that the miRNA target site changes identify tumor cell proliferation and immune cell infiltration to the tumor microenvironment better than the miRNA expression levels or the APA events themselves. Altogether, our computational analyses provide a proof-of-concept demonstration that the miRNA target site information indicates the effect of global APA events with a potential as predictive biomarker.
Keywords: cancer, microRNA, posttranscriptional regulation, tumor heterogeneity
Introduction
The dynamic usage of the messenger RNA (mRNA) 3′-untranslated region (3′-UTR) through alternative polyadenylation (APA) results in transcription of distinct isoforms with shortened or lengthened 3′-UTRs. 3′-UTR lengthening (3′UL) was recently reported to regulate cell senescence [1] that is linked with tumor-suppressive pathways such as cell cycle inhibitors and DNA damage markers [2–4]. 3′-UTR shortening (3′US) was reported widespread in diverse types of human cancers [5]. Further, the reported impact of 3′US on cancer prognosis [5] and on drug sensitivity [6] suggests clinical implications of APA events in human cancer. We recently identified a 3′US trans tumorigenic mechanism [7] that concerns the microRNA (miRNA) target activity [8]. Since 3′US removes miRNA target sites in the 3′-UTRs, genes with shortened 3′UTRs (3′US genes) do not sequester the miRNAs. Then, the miRNAs released from the 3′US genes would be redirected to and repress the other genes that would compete for the miRNAs with the 3′US genes if they did not undergo 3′US events. Since the repressed genes are enriched for tumor suppressors such as the phosphatase and tensin homologue (PTEN), 3′US promotes tumorigenesis in trans. Recently, several studies emerge supporting this trans effect e.g. for PTEN [9] and in the context of alternative splicing [10], another posttranscriptional regulation concerning the miRNA target activity. Further, we also found that this 3′US trans mechanism is involved in the subtype-specific tumor growth of breast cancer [11].
Although global 3′US events remove miRNA target sites in cancer cells, it is challenging to accurately estimate the effect of 3′US events for each miRNA. First, although 3′US removes miRNA target sites in the 3′-UTRs, another type of APA events, 3′UL, can add some of the target sites back. Second, APA events and their associated miRNAs are on many-to-many relationships, making it difficult to pinpoint the miRNAs of which global APA events increase or decrease the target sites. To address these challenges, we developed a mathematical model that estimates the statistical significance of the APA effect on miRNA target sites, probabilistic inference of microRNA target site modification through APA (PRIMATA-APA). Using PRIMATA-APA on the cancer genome atlas (TCGA) breast cancer data [12], we found that the global APA events collectively increase or decrease the target sites of the miRNAs that are known to regulate cancer etiology and treatments. Our results were replicated in a reanalysis of NUDT21 knockdown (KD) data. NUDT21 KD was shown to induce global 3′US events and promote tumorigenesis in vivo and in vitro [13] by removing miRNA target sites to repress tumor suppressor genes [7]. Further, we showed that although the NUDT21 KD in HeLa and the TCGA breast cancer carry a distinct set of 3′US genes, they change the target sites of the common miRNAs (tamoMiRNA), suggesting that the APA-initiated tumorigenesis is attributable to the miRNA target site changes, not the APA events themselves. We further showed that this feature distinguished tumor samples that are subject to high proliferation or high infiltration with immune cells, suggesting this feature as a potential predictive biomarker for immunotherapy with biological implication.
Results
Collective impact of APA events for the trans effect in TCGA breast cancer
To identify genes with APA events (APA genes) based on RNA-seq data, several computational tools have been developed [5, 14–17]. One of the widely used tools, DaPars, identified widespread APA events in diverse cancer data including seven types of TCGA cancer data [5]. The APA events have been further extended and successfully reproduced by others some with experimental validations (e.g. [7, 13, 18], see Methods). DaPars estimates the Percentage of Distal polyA site Usage Index (PDUI) for each gene in tumor versus normal samples and call APA events if PDUI values are different with (1) statistical significance (adjusted P-value <0.05) and (2) absolute difference (∆PDUI <−0.2 for 3′US or >0.2 for 3′UL). The second criterion was employed to select strong cis targets from the widespread APA events.
However, to study the trans effect of the APA events that we recently identified [7], all statistically significant APA genes, regardless of the absolute difference, need to be considered. Previously, we identified that 3′US events remove miRNA target sites to repress trans genes enriched in tumor suppressor genes, thereby contributing to tumorigenesis [7]. In removing miRNA target sites for the trans effect, all significant 3′US genes contribute especially when they are highly expressed. For even more comprehensive understanding, we found that 3′UL genes also need to be considered, since they can add miRNA target sites back to the miRNA target landscape. In this work, we used DaPars on the TCGA breast cancer data (n = 106, see Methods) to find that there are many significant APA genes (adjusted P-value <0.05), either 3′US or 3′UL, without the absolute PDUI difference (−0.2 < ∆PDUI < 0.2) among expressing genes. For example, tumor sample labeled as TCGA-BH-A1FJ carries the most number (3015) of significant APA genes among the TCGA breast cancer data, and the significant APA genes without the absolute PDUI difference account for almost half (49.5%, 1492) of them (Figure 1A). This trend holds true in most of the breast cancer sample pairs (59.8% on average, Figure 1B). Previous studies reported smaller numbers of 3′UL genes than 3′US in TCGA data [5] or cancer cell lines [6]. This was partly because they focused only on those with absolute difference. Considering all significant APA genes in our TCGA breast cancer analysis, we found that 3′UL is as widespread as 3′US. For example, in the tumor sample labeled as TCGA-AC-A2FB, which has the smallest ratio of the APA genes with absolute difference to all significant APA genes, 90.1% of the significant APA genes are 3′UL genes (Supplementary Figure 1A). In 106 TCGA breast tumor-normal sample pairs, average 56.4% of significant APA genes are 3′UL genes (Supplementary Figure 1B), totaling 7265 significant 3′UL genes. Based on the results, we will consider all significant APA genes to study the trans effect.
Figure 1.
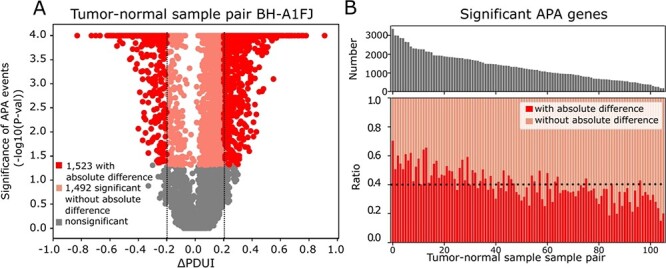
Collective impact of strong and significant APA events. A. Statistical significance of APA genes in a breast tumor-normal sample pair (TCGA-BH-A1FJ) with their ∆PDUI (percentage of PDUI) values (tumor-normal). Since PDUI represents the ratio of isoforms with dUTR, negative ∆PDUI value represent 3′US target genes and positive ∆PDUI value 3′UL genes. Strong APA target genes are in red, significant but not strong ones in pink and not significant ones in gray. B. For 106 breast tumor-normal sample pairs sorted by the number of significant APA target sites, upper panel shows the total number of significant APA genes and the lower panel shows the ratio of the APA genes by whether it is significant but not strong (orange) or strong (red). Black dotted line represents the average ratio of strong APA genes.
Probabilistic inference of miRNA target site modification through APA
To systematically analyze the trans APA effect, focusing on the 3′UL and 3′US genes is challenging for two reasons. First, although 3′US genes remove miRNA target sites in the cell, 3′UL genes add some of the target sites back, complicating the analysis of the collective effect in the presence of global APA events. Second, each 3′US and 3′UL gene will affect the target sites of different sets of miRNAs, further complicating the analysis. To address this challenge, we quantified their collective effect on each miRNA by developing a mathematical model, PRIMATA-APA. Previously, we successfully predicted gene expression changes (R2 = 0.47 for 1548 differentially expressed genes in the TCGA breast tumor samples) by estimating the number of miRNA target sites in the presence of 3′US (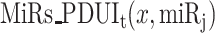 , Eq. 2) [7]. In this work, we extend this formula to estimate the total number of target sites for each miRNA with and without consideration of APA events (Eqs 3 and 4, PRIMATA-APA). Based on this estimation, PRIMATA-APA estimates how significantly the APA events increase or decrease the target sites for each miRNA (see Methods). In each of the 70 breast tumor-matched normal sample pairs out of 106, for which miRNA expression information was available, we ran PRIMATA-APA for each of 588 moderately expressed miRNAs (1 < average fragments per million mapped fragments (FPM) < 100, Figure 2A). Summarizing the result by sample pair, PRIMATA-APA identifies significant target site changes (false discovery rate (FDR) < 0.01) (tumor versus normal) for more than 100 miRNAs in 39 (55.7%) of the 70 breast tumor samples, (Figure 2B), showing the global impact of APA events on the miRNA targeting landscape. Summarizing the result by miRNA, the number of miRNA target sites increased and decreased shows a significant negative correlation across the sample pairs (P = 0.006, Figure 2C) despite the small sample size (n = 70). This result suggests that the global APA events modify the miRNA target sites in a nonrandom fashion, either increasing or decreasing altogether in each tumor sample.
, Eq. 2) [7]. In this work, we extend this formula to estimate the total number of target sites for each miRNA with and without consideration of APA events (Eqs 3 and 4, PRIMATA-APA). Based on this estimation, PRIMATA-APA estimates how significantly the APA events increase or decrease the target sites for each miRNA (see Methods). In each of the 70 breast tumor-matched normal sample pairs out of 106, for which miRNA expression information was available, we ran PRIMATA-APA for each of 588 moderately expressed miRNAs (1 < average fragments per million mapped fragments (FPM) < 100, Figure 2A). Summarizing the result by sample pair, PRIMATA-APA identifies significant target site changes (false discovery rate (FDR) < 0.01) (tumor versus normal) for more than 100 miRNAs in 39 (55.7%) of the 70 breast tumor samples, (Figure 2B), showing the global impact of APA events on the miRNA targeting landscape. Summarizing the result by miRNA, the number of miRNA target sites increased and decreased shows a significant negative correlation across the sample pairs (P = 0.006, Figure 2C) despite the small sample size (n = 70). This result suggests that the global APA events modify the miRNA target sites in a nonrandom fashion, either increasing or decreasing altogether in each tumor sample.
Figure 2.
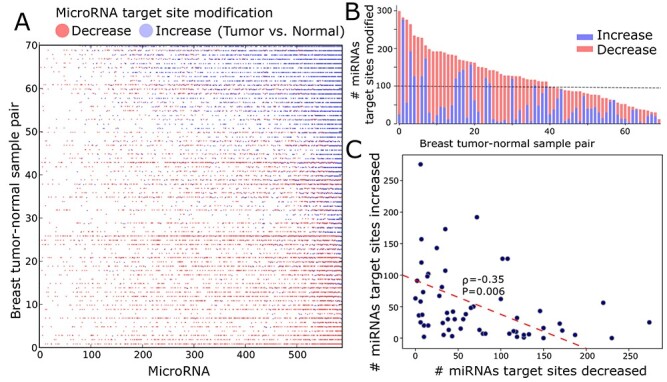
Tumor-specific APA-derived miRNA target site changes. A. The heatmap shows tumor-normal samples (row) where the total number of target sites for each miRNA (column) is increased (blue) or decreased (red) due to APA. Not significant changes or no changes are not colored. Samples are sorted by the number of increased miRNA target site modification. B. The total number of miRNA target site changes, either increased (blue) or decreased (red) due to APA, in breast tumor-normal samples pair sorted by the target site number changes per sample pair. C. Number of miRNAs of which target sites are increased (y-axis) or decreased (x-axis) in each tumor-normal sample. The red dotted line represents linear least-squares regression.
APA changes the number of target sites of miRNAs associated with cancer
To identify miRNAs of which APA events significantly change the number of target sites, we examined the number of tumor samples in which the target sites are significantly increased or decreased (Figure 3A). The result shows that the APA events focus on changing the target sites of particular miRNAs (P-value = 1.12e−28). We selected top half (289) of the miRNAs by the number of samples in which APA events significantly changed the target site numbers [target sites modified miRNA (tamoMiRNA), Supplementary Table 1]. Further, we found that tamoMiRNAs are significantly more enriched in the miRNAs known for biological roles in cancer development and treatment than the other miRNAs (P = 5.8e−5, Figure 3B and Supplementary Table 1). Specifically, tamoMiRNAs are enriched in the miRNAs that are dysregulated in breast cancer with clinical and biological implications [19], regulating diverse mechanisms for breast cancer [20, 21], regulatory elements in either adaptive or innate immune system [22] or potential prognostic and predictive biomarkers identified for breast cancer [21]. For example, APA events significantly change the target sites for miR-221/222 in 61 TCGA breast tumor samples (increased in 36 samples and decreased in 25 samples), ranked top fourth among all tamoMiRNAs. miR-221/222 has been reported to be a crucial oncoMiRNA by targeting the cyclin-dependent kinase inhibitor p27Kip1 and accelerating cell proliferation [23–26]. Further, miR-221/222 inhibits estrogen receptor α and is associated with tamoxifen and fulvestrant resistance of breast cancer cells [27–29]. Among 43 tamoMiRNAs found in the categories, 31 (72.1%) occur only in one of the categories (Supplementary Figure 2), confirming that the high enrichment of tamoMiRNAs to the multiple categories reflects their important roles in tumor, not redundancy in data collection. Also, we estimated conservation score (PhyloP [30], 46 way Placental) of 202 tamoMiRNAs and 191 other miRNAs for which miRbase [31] uniquely curated the genomic locations. TamoMiRNAs have significantly (P = 7.19e−5) larger conservation scores than the other miRNAs (Figure 3C), implicating their functional significance. Altogether, these results indicate that the global APA events in breast cancer are associated with the miRNAs that are evolutionary conserved and functionally important for cancer etiology and treatments.
Figure 3.
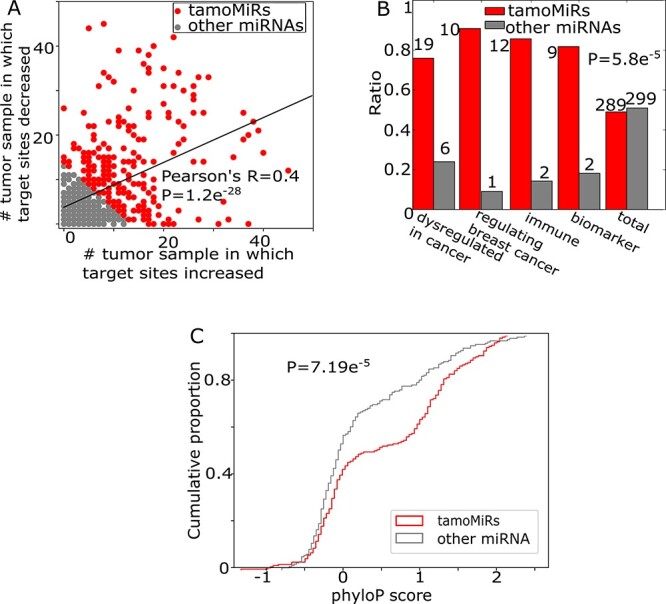
APA modifies miRNA target sites associated with cancer. A. The number of tumor-normal samples between which target sites for each miRNA are increased (x-axis) or decreased (y-axis). For further analyses, we dichotomize miRNAs by the amount of target site changes into tamo- (red) and the other (gray) miRNAs (Supplementary Table 1). B. Number of cancer-related miRNAs in tamo- (red) and the other (gray) miRNAs. C. The distribution of phyloP conservation score for 202 tamo- and 191 the other miRNAs.
APA changes the number of miRNA target sites to effectively regulate biological processes
To further understand the role of the miRNA target site changes, we analyzed how efficiently the miRNAs would regulate important biological processes. First, we identified GO terms enriched for tamoMiRNAs using MiEAA [32]. Probably due to the many-to-many relationships between miRNAs and the target genes [33–35], inputting all tamoMiRNAs to the MiEAA web server returns mostly under-represented terms. So, we ran MiEAA on 102 tamoMiRNAs (with the greatest number of samples in which changed) and 104 other miRNAs (with the least number of samples in which changed). A total of 125 biological terms are significantly (FDR < 0.01) enriched for tamoMiRNAs, whereas only one biological term is significantly enriched for the other miRNAs (Supplementary Table 2). The significant bias of the number of enriched biological terms to tamoMiRNAs (P-value = 5.0 × 10−5) suggests that APA changes the miRNA target site numbers for effective regulation of biological functions. Additionally, compared to the other miRNAs, tamoMiRNAs are exclusively enriched for pathways with keyword ‘growth factor (GF)’, ‘signaling’ and ‘circadian’ (Figure 4A and Supplementary Table 2), which are essential for tumor initiation and progression [36].
Figure 4.
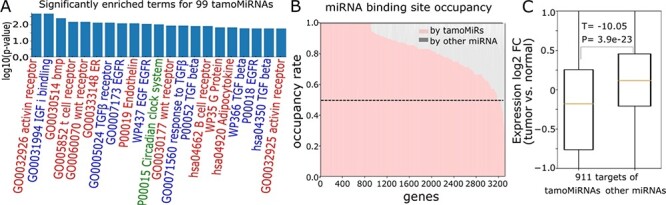
TamoMiRNAs effectively regulate biological processes. A. Cancer-associated pathways enriched for 99 tamoMiRNAs with their enrichment P-values (red for ‘signaling’, blue for ‘GF’ and green for ‘circadian’). B. Number of target sites for tamoMiRs and the other miRNAs in the genes with more than five target sites. C. Expression fold change (log2 tumor versus normal) of 911 genes that are targets of tamoMiRs and other miRNAs.
Then, we compared the tamoMiRNA target genes with those of the other miRNAs. Among 3318 expressed genes in the breast tumor data that are likely controlled by miRNAs (>5 miRNA target sites), 3177 genes (95.7%) have more target sites for tamoMiRNAs (Figure 4B). Further, 911 of 3177 (27.4%) genes have target sites only for tamoMiRNAs in their 3′-UTRs. Although the expression fold change (tumor versus normal) does not differ between tamoMiRNAs and the other miRNAs (P = 0.1, Supplementary Figure 3), 911 genes targeted only by tamoMiRNAs are significantly more down-regulated in tumor (P = 3.9e−23) than the same number of genes affected by the other miRNAs (Figure 4C). Collectively, the results suggest that APA focuses on changing the target sites of the miRNAs that effectively regulate the genes in the pathways implicated for tumorigenesis.
NUDT21 knockdown induces a different set of APA genes that modifies target sites of the common miRNAs
Previously, knocking down NUDT21 in HeLa cells demonstrated the causal role of 3′US for tumorigenesis in a miRNA-dependent fashion [13]. In a follow-up study, we found that the tumorigenic effect involves miRNA target site changes for particular genes including tumor suppressors, such as PTEN and PHF6 [7]. We reanalyzed this experiment data to test if the miRNA target site change can be considered as an independent feature associated with tumorigenesis without association to particular genes. First, in two replicate data of NUDT21 KD versus wild type (WT) HeLa cells, we identified global significant APA genes (FDR < 0.05) without absolute difference (615 3′UL and 2847 3′US, 0.2 > ∆PDUI > −0.2, Figure 5A). To check the consistency in the miRNA target site number changes between the replicates, we ran PRIMATA-APA on 383 moderately expressed miRNAs (1 < average FPM < 100) in two random pairs of NUDT21 KD versus WT. Between the pairs, we found that the increase/decrease call for each miRNA agree using Cohen’s κ (97.4% of agreement, P < 10e−16 using R package ‘irr’). With the high consistence of PRIMATA-APA calls between the pairs, we ran PRIMATA-APA on the averaged expression of two WTs and two NUDT21 KD (Supplementary Table 3). Then, we ranked the miRNAs based on the χ2 value that indicates the degree of APA-derived target site number change (see Methods). Then, we call the top half of the miRNAs (191) tamoMiRNAs as before. It is interesting that miR-3187-3p, which was extensively validated to promote tumorigenesis in the NUDT21 KD [7] experiment, is a tamoMiRNA [7] for NUDT21 KD.
Figure 5.
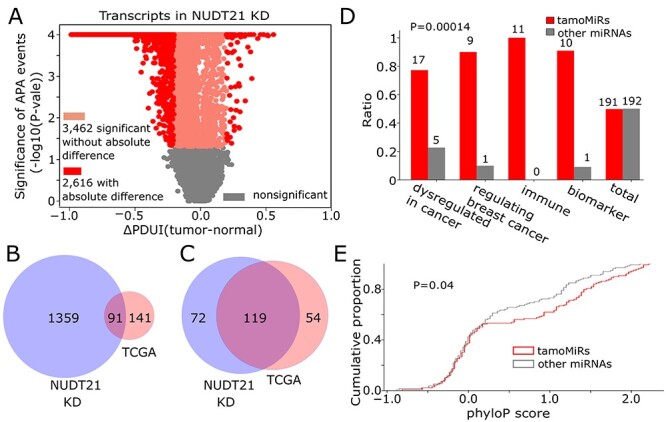
KD of NUDT21, an upstream regulator of global 3′US leading to tumorigenesis, induces a different set of APA genes that changes target sites of the common miRNAs. A. Statistical significance of APA genes in NUDT21 KD experiment data with their ∆PDUI values (tumor-normal). Overlap of B. 3′US genes and C. tamoMiRNAs between TCGA breast cancer data and NUDT21 KD data based on PRIMATA-APA. D. Number of cancer-related miRNAs in 191 tamo- (red) and the other (gray) 192 miRNAs. E. The distribution of phyloP conservation score for 139 tamo- and the other 134 miRNAs.
Since NUDT21 KD and the TCGA breast cancer data represent different 3′US-mediated tumorigenic model, it makes sense that they are identified with different sets of 3′US genes (14-fold less in common than unique to NUDT21 KD, Figure 5B). However, they share a significant number of tamoMiRNAs (more in common than unique to either NUDT21 KD or TCGA, P = 1.16 × 10−11, Figure 5C). It follows that the tamoMiRs for NUDT21 KD data also replicates the high enrichment of the miRNAs for cancer etiology and treatments (P = 0.00014, Figure 5D) with high evolutional conservation (P = 0.04, Figure 5E). The results suggest that APA-mediated miRNA target site number changes, not the APA events themselves, is common to two distinct tumorigenic models, NUDT21 KD and TCGA breast cancer.
The miRNA target landscape distinguishes tumor samples of different immune evasion activity
To understand biological implication of miRNA target site modification with a potential clinical use, we tested the association of the APA-mediated miRNA target site changes with tumor samples’ immune evasion activity. Immune evasion is an important cancer hallmark and has long been recognized as a fundamental process in tumor formation and progression [37, 38]. Although the mechanisms controlling immune infiltration are not well-understood [39], several research groups have identified molecular indicators for immune evasion activities. For example, expression of cell proliferation markers (proliferation signature) and of cytotoxic infiltrating immune cells (immune signature) were identified and shown associated with immune evasion and reduced response to immunotherapy [40]. Here, we showed that the miRNAs shown for tumor proliferation and immune evasion are associated with proliferation and immune signature through the target site changes. From 11 miRNAs that are shown in literature [20] to regulate breast cancer (Figure 3B), we further selected 6 miRNAs that were shown to directly promote cancer (oncogenic miRNAs) and ranked the 70 TCGA breast tumor samples by the PRIMATA-APA increase/decrease call for the miRNAs. We found that the tumor samples with increase calls (blue points in Figure 6A, P-value = 0.03) carry significantly lower proliferation scores. Similarly, the samples with decrease calls for the miRNAs shown to regulate the immune system [22] (immune miRNAs) significantly (P-value = 0.01) select tumor samples with high immune score (Figure 6D). On the other hand, the expression information of such miRNAs does not distinguish tumor samples with different proliferation score (P-value = 0.7, Figure 6B), although it performs as well as the miRNA target site information for immune signature (P-value = 0.01, Figure 6E). Moreover, the number of APA genes that change the target site numbers of the oncogenic and immune miRNAs cannot distinguish the patients (P-value = 0.9, Figure 6C and P-value = 0.8, Figure 6F). Altogether, the results show that the significant separation of tumor samples in immune and proliferation signature score (Figure 6A and B) involves such miRNAs under the trans APA effect.
Figure 6.
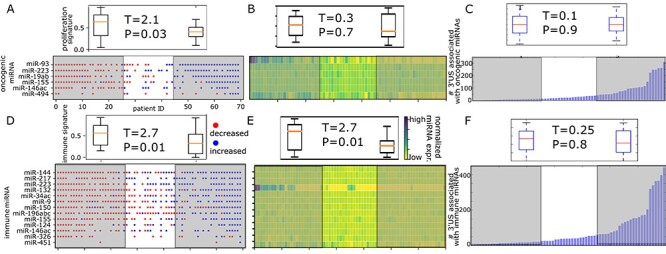
The miRNA target landscape distinguishes tumor samples of different immune activity. A total of 70 TCGA breast tumors ranked by the followings. A. The changes in the decrease (red) or increase (blue) of oncogenic miRNAs. B. The average of normalized miRNA expression of oncogenic miRNAs. C. The total number of APA genes removing/adding target sites of oncogenic miRNAs (bottom panel). The distribution of proliferation signature score of the 25 tumor samples in the left/right-most of the rank (top panel). D. The changes in the decrease (red) or increase (blue) of immune miRNAs. E. The average of normalized miRNA expression of immune miRNAs. F. The total number of APA genes removing/adding target sites of immune miRNAs (bottom panel). The distribution of immune signature score of the 25 tumor samples in the left/right-most of the rank (top panel). Test statistics and the P-values are based on t-test for two independent samples.
Discussion
Since the discovery of miRNAs in the earlier 1990s, much progress has been made in understanding how miRNAs are produced within cells and how they are involved in various physiological and pathological contexts. To understand miRNA binding efficiency, previous works have used miRNA’s expression information almost as the sole molecular feature. For example, miRNAs up-regulated in tumor were considered to be oncogenic miRNAs and those down-regulated were tumor-suppressive [41, 42]. However, as miRNAs play roles majorly by binding their target genes [43], a comprehensive understanding of miRNAs’ function should be based not only on their expression levels but also on the targeting landscape. Considering the targeting landscape becomes especially important in studying cancer where the widespread APA globally changes miRNA target sites. In this work, we characterized the amount of miRNA target sites as a novel molecular feature that reflects the effect of widespread APA events on the miRNA targeting landscape. To systematically understand the role of this molecular feature in TCGA breast cancer, we develop a mathematical model, PRIMATA-APA. Running PRIMATA-APA on TCGA breast cancer data and NUDT21 KD data, we found that considering miRNA target site change brings a novel understanding into the tumorigenic mechanism of the widespread APA events in association with miRNA binding efficiency.
Based on the understanding, our work sheds novel insights into the development of therapeutic miRNAs. TamoMiRNAs are highly enriched for miRNAs that are known to regulate important biological processes in cancer (Figure 3B and 4A). This result suggests that, when APA events are shown to affect tumorigenesis [7] and associated with prognosis [5] and treatment outcomes [18], the APA events are expected to play such roles by increasing/decreasing the miRNA target sites. Further, we found that the trans APA effect of the validated miRNA biomarkers identifies tumor samples of different tumor cell proliferation and immune evasion activity (Figure 6A, D). This result provides a clinical implication of therapeutic miRNA agents, miRNA mimics or anti-miRNAs. For example, the target site increase for the oncogenic miRNAs is associated with lower tumor cell proliferation in cancer patients (Figure 6A). Since the target site increase will make the targeting less efficient, anti-miRNAs for the miRNAs are expected to lower tumor cell proliferation status. In the same sense, since the target site decrease for the immune miRNAs is associated with higher immune infiltration (Figure 6D), miRNA mimics of the immune miRNAs is expected to reproduce the effect and induce immune infiltration. Altogether, we showed that widespread APA events globally change miRNA target sites and considering the target site change helps better understand the functional role of miRNAs in cancer biology.
Methods
TCGA breast tumor RNA-seq and miRNA-seq data
Quantified gene expression files (RNASeqV1) for primary breast tumors and their matching solid normal samples were downloaded from TCGA data portal [44]. We used 106 breast tumor samples that have matched normal tissues. A number of 10 868 expressed RefSeq genes (fragments per kilobase million (FPKM) ≥ 1 in >80% of all samples) were selected for downstream analyses. To better quantify gene expression in the presence of 3′US, we only used coding regions (CDS). Exon and CDS annotation for TCGA data and miRNA expressions (syn1445790) were downloaded from Sage Bionetworks’ Synapse database.
Linking miRNAs and the targets in stoichiometry
Predicted miRNA target sites were obtained from TargetScanHuman version 6.2 [45]. Only those with a preferentially conserved targeting score more than 0 were used [5]. Experimentally validated miRNA-target sites were obtained from TarBase version 5.0 [46], miRecords version 4 [47] and miRTarBase version 4.5 [48]. The target sites found in indirect studies such as microarray experiments and high-throughput proteomics measurements were filtered out [49]. Another source is the miRNA target atlas composed of public AGO-CLIP data [50] with significant target sites (q-value <0.05). The predicted and validated target site information was then combined to use in this study. Further, to model the potential targeting of miRNAs on mRNA, we will take into account only those that were considered for that purpose [7, 51, 52] (expressed miRNAs (>1 and <100 FPM on average) and the expressed genes (avg. FPKM > 1)).
Detection of miRNA target sites removed/added by APA events
PRIMATA-APA uses DaPars to identify APA events [5]. We chose DaPars method because its performance has been validated not only within the DaPars paper but also in an independent recent study. Within the DaPars paper, multiple lines of evidence were presented to demonstrate that DaPars indeed identified APA events in the TCGA data. First, 51% of the DaPars predictions are within 50 bp of the annotated APAs in the gene models compiled from NCBI Reference Sequence database (RefSeq), Ensembl genome database (Ensembl), UCSC genome browser and polyA database (polyA_DB [53]). Second, in the upstream (−50 nt) of the predicted APA sites, MEME motif enrichment analysis [54] successfully identified canonical polyA signal AATAAA.
An independent recent study [55] assessed the performance of many APA detection tools, such as PHMM [56], GETUTR [57], ChangePoint [17], EBChangePoint [58], IsoSCM [59], DaPars [5], QAPA [14], APATrap [60] and TAPAS [61] using diverse simulated and biological data. The study concluded that DaPars is among the top performers with TAPAS and APAtrap according to the prediction precision or they are under the receiver operating characteristics (ROC) curves.
To identify the miRNA target sites removed or added by APA events, we map the poly (A) site (PAS) most proximal to the stop codon predicted by DaPars to the assignment of miRNA target sites collected above. With the PDUI values returned from DaPars running, further analyses were performed.
Probabilistic inference of miRNA target site modification through APA
To estimate the total number of target sites for each miRNA, we consider all genes with the target sites (based on TargetScan prediction) and the expression information of the genes (estimated in the data). Suppose transcript 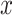 has a constitutive proximal 3′-UTR (pUTR) and a distal 3′-UTR (dUTR). Previously [7], we defined the amount of target sites for miRNA miRj in all copies of transcript x based on the gene model predictions annotated in TargetScan.
has a constitutive proximal 3′-UTR (pUTR) and a distal 3′-UTR (dUTR). Previously [7], we defined the amount of target sites for miRNA miRj in all copies of transcript x based on the gene model predictions annotated in TargetScan.
 |
(1) |
where pUTR (x, miRj) and dUTR (x, miRj) are the numbers of 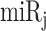 target sites in pUTR and dUTR of x. PDUIt (x) is the Percentage of dUTR Usage Index [5] of x and CDSt (x) is the expression level of x in a tumor sample. Note that
target sites in pUTR and dUTR of x. PDUIt (x) is the Percentage of dUTR Usage Index [5] of x and CDSt (x) is the expression level of x in a tumor sample. Note that 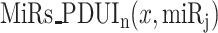 can be calculated for a normal sample with PDUIn (x) and CDSn (x). If APA-derived miRNA target site modification is not considered, the amount of target sites for
can be calculated for a normal sample with PDUIn (x) and CDSn (x). If APA-derived miRNA target site modification is not considered, the amount of target sites for 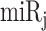 in all copies of transcript x would be calculated as follows:
in all copies of transcript x would be calculated as follows:
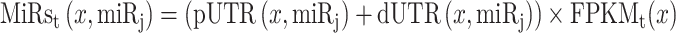 |
(2) |
In this manuscript, PRIMATA-APA defines the total number of miRNA target sites with and without consideration of APA events as below.
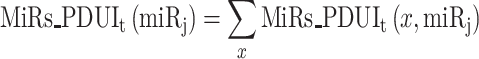 |
(3) |
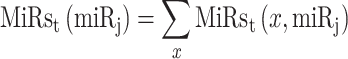 |
(4) |
With 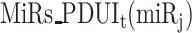 ,
, 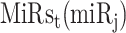 ,
, 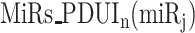 and
and 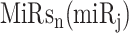 in the contingency table, PRIMATA-APA estimates significance of target site modifications for
in the contingency table, PRIMATA-APA estimates significance of target site modifications for 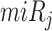 by testing nonrandom association in tumor and normal states (using χ2 test), followed by FDR control using FowardStop [62] (FDR < 0.01). Note that we have run this model on the transcript level (isoforms). Then, the transcript level estimates were combined on the gene level for interpretation. While this model explicitly takes miRNA target site and gene expression information into account, miRNA expression information is implicitly considered as we ran this model only for expressed miRNAs defined above.
by testing nonrandom association in tumor and normal states (using χ2 test), followed by FDR control using FowardStop [62] (FDR < 0.01). Note that we have run this model on the transcript level (isoforms). Then, the transcript level estimates were combined on the gene level for interpretation. While this model explicitly takes miRNA target site and gene expression information into account, miRNA expression information is implicitly considered as we ran this model only for expressed miRNAs defined above.
Collection of miRNAs known for biological roles
MiRNAs dysregulated in breast cancer with clinical and biological implications are taken from the section MECHANISMS OF MIRNA DYSREGULATION IN CANCER AND SIGNIFICANCE OF THE ALTERED MIRNA EXPRESSION IN TUMORS in [19]. MiRNAs regulating diverse mechanisms for breast cancer are taken from Table 2 The miRNAs associated with tumor development in breast cancer from [9] and Table 1 List of major oncogenic miRNAs in breast cancer and Table 2 List of major tumor-suppressive miRNAs in breast cancer from [21]. Regulatory miRNAs in either adaptive or innate immune system are taken from Figure 4 miRNAs that regulate innate immune cell development and function from and Figure 5 miRNAs that regulate adaptive immune cell development and function from [21]. Potential prognostic and predictive miRNA biomarkers identified for breast cancer are taken from Table 4 List of major diagnostic miRNA signatures for the early diagnosis of breast cancer, Table 7 Predictive miRNAs–miRNAs involved in response (sensitivity/resistance) to conventional breast cancer therapeutic strategies, Table 8 List of positive prognostic miRNA signatures in breast cancer, Table 9 List of negative prognostic miRNA signatures in breast cancer from [21].
Selection of tamoMiRNAs and the other miRNAs
To select 102 tamoMiRNAs and 104 other miRNAs, we used the consistent criteria described as follows. We sorted the miRNAs by the number of samples in which the number of the target sites are significantly modified (increased or decreased) by APA events. When there are multiple miRNAs with the same number of such samples, they are taken together. For example, 93th to 102th miRNAs in the ranked list were modified in the same number (24) of such samples (Supplementary Table 1) that they need to be considered together. In that way, 102 tamoMiRNAs were the minimum number of miRNAs that produced meaningful results in the MiEAA analysis. Further, 104 other miRNAs were the minimum number of miRNAs on the other side of the ranked list that are closest to 102.
Author Contributions
H.J.P and S.K. conceived the project, designed the experiments and performed the data analysis. Y.B. and Z.F. performed the subtype analysis. H.J.P and S.K. wrote the manuscript with input from B.D. and G.C.T.
Availability of data and materials
The open source PRIMATA-APA program (version 0.9.2) is freely available at https://github.com/thejustpark/PRIMATA-APA/ with necessary example data for this analysis.
Key Points
APA is widespread in diverse types of cancer, including breast cancer. Although it has been shown associated with prognosis and drug sensitivity in cancer, it is not widely known how it plays roles.
Although our recent finding that APA plays roles in trans is limited to tumor suppressor genes, we develop a mathematical model to elucidate the systematic roles on miRNA target site landscape.
Using the model on breast cancer data, we identified that particular miRNAs’ target sites are extensively modified by APA events and that some of the miRNAs have been shown important in tumor biology and treatment.
With the results, our computational analyses provide a proof-of-concept demonstration that the miRNA target site information indicates the effect of global APA events with a potential as predictive biomarker.
Supplementary Material
Acknowledgments
This research was supported in part by the University of Pittsburgh Center for Research Computing through the resources provided.
Soyeon Kim is a postdoctoral associate in Department of Pediatrics, University of Pittsburgh Medical Center and in Division of Pulmonary Medicine, Children’s Hospital of Pittsburgh of UPMC. She studies statistical aspects of posttranscriptional dynamics in cancer.
Yulong Bai is a PhD student in Department of Human Genetics in the Graduate School of Public Health, University of Pittsburgh. He studies alternative polyadenylation dynamics in physiological and pathological conditions.
Zhenjiang Fan is a PhD student in Department of Computer Science, University of Pittsburgh. He studies biological network analysis techniques.
Brenda Diergaarde is an associate professor in Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh. She studies epidemiological models for cancer patients.
George C. Tseng is a professor in Department of Biostatistics, Graduate School of Public Health, University of Pittsburgh. He studies statistical modeling of biological and clinical data.
Hyun Jung Park is an assistant professor in Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh. He studies computational and statistical aspects of posttranscriptional regulation in cancer.
Contributor Information
Soyeon Kim, Department of Pediatrics, University of Pittsburgh Medical Center and in Division of Pulmonary Medicine, Children’s Hospital of Pittsburgh of UPMC.
YuLong Bai, Department of Human Genetics in the Graduate School of Public Health, University of Pittsburgh.
Zhenjiang Fan, Department of Computer Science, University of Pittsburgh.
Brenda Diergaarde, Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh.
George C Tseng, Department of Biostatistics, Graduate School of Public Health, University of Pittsburgh.
Hyun Jung Park, Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh.
Funding
The National Institutes of Health through Grant Number UL1TR001857, University of Pittsburgh, Department of Human Genetics – Joan Gollin Gaines Cancer Research Fund, University of Pittsburgh, Department of Human Genetics – startup funding, all awarded to H.J.P. UPMC Hillman Cancer Center Biostatistics Shared Resource(P30CA047904).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1. Chen M, Lyu G, Han M, et al. 3′ UTR lengthening as a novel mechanism in regulating cellular senescence. Genome Res 2018;28:285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995;92:9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Busuttil RA, Rubio M, Dollé MET, et al. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell 2003;2:287–94. [DOI] [PubMed] [Google Scholar]
- 4. López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell 2013;153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xia Z, Donehower LA, Cooper TA, et al. Dynamic analyses of alternative polyadenylation from RNA- seq reveal landscape of 3′ UTR usage across 7 tumor types. Nat Commun 2014;1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiang Y, Ye Y, Lou Y, et al. Comprehensive characterization of alternative polyadenylation in human cancer. Journal of the National Cancer Institute 2018;110:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park HJ, Ji P, Kim S, et al. 3′ UTR shortening represses tumor-suppressor genes in trans by disrupting ceRNA crosstalk. Nat Genet 2018;50:783–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park HJ, Kim S, Li W. Model-based analysis of competing- endogenous pathways ( MACPath ) in human cancers. PLoS Comput Biol 2018;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thivierge C, Tseng H-W, Mayya VK, et al. Alternative polyadenylation confers Pten mRNAs stability and resistance to microRNAs. Nucleic Acids Res 2018;46:10340–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han S, Kim D, Shivakumar M, et al. The effects of alternative splicing on miRNA binding sites in bladder cancer. PLoS One 2018;13:e0190708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhenjiang F, Kim S, Diergaarde B, et al. 3′-UTR shortening disrupts ceRNA crosstalk of housekeeping genes resulting in subtype-specific breast cancer development. bioRxiv 2019;601526. [Google Scholar]
- 12. Network TCGA. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Masamha CP, Xia Z, Yang J, et al. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 2014;510:412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ha KCH, Blencowe BJ, Morris Q. QAPA: a new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biology 2018;45:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ye C, Long Y, Ji G, et al. APAtrap: identification and quantification of alternative polyadenylation sites from RNA-seq data. Bioinformatics 2018;34:1841–9. [DOI] [PubMed] [Google Scholar]
- 16. Huang Z, Teeling EC. ExUTR: a novel pipeline for large-scale prediction of 3′-UTR sequences from NGS data. BMC Genomics 2017;18:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang W, Wei Z, Li H. A change-point model for identifying 3′UTR switching by next-generation RNA sequencing. Bioinformatics 2014;30:2162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiang Y, Ye Y, Lou Y, et al. Comprehensive characterization of alternative Polyadenylation in human cancer. JNCI J Natl Cancer Inst 2017;110:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther 2016;1:15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kurozumi S, Yamaguchi Y, Kurosumi M, et al. Recent trends in microRNA research into breast cancer with particular focus on the associations between microRNAs and intrinsic subtypes. 2016;62:15–24. [DOI] [PubMed] [Google Scholar]
- 21. Schooneveld E, Wildiers H, Vergote I, et al. Dysregulation of microRNAs in breast cancer and their potential role as prognostic and predictive biomarkers in patient management. Breast Cancer Res 2015;17:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol 2016;16:279–94. [DOI] [PubMed] [Google Scholar]
- 23. Visone R, Russo L, Pallante P, et al. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr Relat Cancer 2007;14:791–8. [DOI] [PubMed] [Google Scholar]
- 24. Galardi S, Mercatelli N, Giorda E, et al. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. The Journal Of Biological Chemistry 2007;282:23716–24. [DOI] [PubMed] [Google Scholar]
- 25. Gillies JK, Lorimer IAJ. Regulation of p27Kip1 by miRNA 221/222 in Glioblastoma. Cell Cycle 2007;6:2005–9. [DOI] [PubMed] [Google Scholar]
- 26. Sage C, Nagel R, Egan DA, et al. Regulation of the p27Kip1 tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J 2007;26:3699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wei Y, Lai X, Yu S, et al. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res Treat 2014;147:423–31. [DOI] [PubMed] [Google Scholar]
- 28. Gan R, Yang Y, Yang X, et al. Downregulation of miR-221/222 enhances sensitivity of breast cancer cells to tamoxifen through upregulation of TIMP3. Cancer Gene Ther 2014;21:290–6. [DOI] [PubMed] [Google Scholar]
- 29. Rao X, Di Leva G, Li M, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene 2011;30:1082–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pollard KS, Hubisz MJ, Rosenbloom KR, et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014;42:D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Backes C, Khaleeq QT, Meese E, et al. MiEAA: MicroRNA enrichment analysis and annotation. Nucleic Acids Res 2016;44:W110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ooi CH, Oh HK, Wang HZA, et al. A densely interconnected genome-wide network of micrornas and oncogenic pathways revealed using gene expression signatures. PLoS Genet 2011;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mavrakis KJ, Van Der Meulen J, Wolfe AL, et al. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat Genet 2011;43:673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hashimoto Y, Akiyama Y, Yuasa Y. Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS One 2013;8:e62589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018;173:321–37e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol 2002;3:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013;14:1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rooney MS, Shukla SA, Wu CJ, et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davoli T, Uno H, Wooten EC, et al. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017;355(80):eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shalaby T, Fiaschetti G, Baumgartner M, et al. Significance and therapeutic value of miRNAs in embryonal neural tumors. Molecules 2014;19:5821–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gambari R, Brognara E, Spandidos DA, et al. Targeting oncomiRNAs and mimicking tumor suppressor miRNAs: Ew trends in the development of miRNA therapeutic strategies in oncology (review). Int J Oncol 2016;49:5–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 2019;20:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Goldman M, Craft B, Swatloski T, et al. The UCSC cancer genomics browser: update 2013. Nucleic Acids Res 2013;41:D949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120:15–20. [DOI] [PubMed] [Google Scholar]
- 46. Papadopoulos GL, Reczko M, V a S, et al. The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res 2009;37:D155–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xiao F, Zuo Z, Cai G, et al. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 2009;37:105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hsu S-D, Tseng Y-T, Shrestha S, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res 2014;42:D78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dvinge H, Git A, Gräf S, et al. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature 2013;497:378–82. [DOI] [PubMed] [Google Scholar]
- 50. Hamilton MP, Rajapakshe K, Hartig SM, et al. Identification of a pan-cancer oncogenic microRNA superfamily anchored by a central core seed motif. Nat Commun 2013;4:2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ala U, F a K, Bosia C, et al. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc Natl Acad Sci U S A 2013;110:7154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sumazin P, Yang X, Chiu H-S, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 2011;147:370–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee JY, Yeh I, Park JY, et al. PolyA_DB 2: mRNA polyadenylation sites in vertebrate genes. Nucleic Acids Res 2007;35:165–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bailey TL, Boden M, Buske FA, et al. MEME suite: tools for motif discovery and searching. Nucleic Acids Res 2009;37:202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen M, Ji G, Fu H, et al. A survey on identification and quantification of alternative polyadenylation sites from RNA-seq data. Brief Bioinform 2019;21:1261–76. [DOI] [PubMed] [Google Scholar]
- 56. Lu J, Bushel PR. Dynamic expression of 3′ UTRs revealed by Poisson hidden Markov modeling of RNA-Seq : implications in gene expression pro fi ling. Gene 2013;527:616–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim M, You B, Nam J. Global estimation of the 3′ untranslated region landscape using RNA sequencing. Methods 2015;83:111–7. [DOI] [PubMed] [Google Scholar]
- 58. Zhang J, Wei Z. An empirical Bayes change-point model for identifying 3′ and 5′ alternative splicing by next-generation RNA sequencing. Bioinformatics 2016;32:1823–31. [DOI] [PubMed] [Google Scholar]
- 59. Shenker SOL, Miura P, Sanfilippo P, et al. IsoSCM : improved and alternative 3′ UTR annotation using multiple change-point inference. Bioinformatics 2014;14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ye C, Long Y, Ji G, et al. APAtrap: identification and quantification of alternative polyadenylation sites from RNA-seq data. [DOI] [PubMed]
- 61. Arefeen A, Liu J, Xiao X, et al. TAPAS: tool for alternative polyadenylation site analysis. Bioinformatics 2018;34:2521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. G’Sell MG, Wager S, Chouldechova A, et al. Sequential selection procedures and false discovery rate control. J R Statist Soc B 2016;78:423–44. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The open source PRIMATA-APA program (version 0.9.2) is freely available at https://github.com/thejustpark/PRIMATA-APA/ with necessary example data for this analysis.
Key Points
APA is widespread in diverse types of cancer, including breast cancer. Although it has been shown associated with prognosis and drug sensitivity in cancer, it is not widely known how it plays roles.
Although our recent finding that APA plays roles in trans is limited to tumor suppressor genes, we develop a mathematical model to elucidate the systematic roles on miRNA target site landscape.
Using the model on breast cancer data, we identified that particular miRNAs’ target sites are extensively modified by APA events and that some of the miRNAs have been shown important in tumor biology and treatment.
With the results, our computational analyses provide a proof-of-concept demonstration that the miRNA target site information indicates the effect of global APA events with a potential as predictive biomarker.